Transcriber's Note:
Every effort has been made to replicate this text as faithfully as possible. Some changes have been made. They are listed at the end of the text.
DISSERTATION
SUBMITTED IN PARTIAL FULFILLMENT OF THE REQUIREMENTS
FOR THE DEGREE OF DOCTOR OF
PHILOSOPHY IN THE FACULTY OF PURE
SCIENCE OF COLUMBIA UNIVERSITY
BY
YÜ-GWAN CHEN, M. A.
NEW YORK CITY
1922
The following research was undertaken at the suggestion of Professor Marston Taylor Bogert to whose interest and advice this work owes whatever merit it may possess.
Y. G. CHEN.
1. What was attempted?
Attempt was made to study organic selenium compounds of the heterocyclic series in reference to those properties leading to tinctorial and pharmaceutical possibilities.
2. What were the methods of attack?
(a) Organic selenium compounds were reviewed and their properties examined critically with those of allied compounds.
(b) Some new heterocyclic compounds of selenium were studied and their characteristic properties more closely examined along the desired line.
3. In how far were the attempts successful?
The literature was reviewed and classified with reference to the properties under consideration, and new selenium organic compounds were prepared and studied which it is hoped may throw some additional light upon the problem.
4. What contribution actually new to the science of chemistry has been made?
(a) Compounds newly made have been shown to exhibit a distinct tinctorial value in comparison with their analogues.
(b) They have been shown to be chemically easier to handle than the corresponding sulphur compounds.
(c) Selenium, in the nucleus of cyclic compounds, has been shown to be instrumental for a positive coloration at least equal to the —NH— or —S— groupings. The selenocarbonyl, :C:Se, has been shown to be a more powerful chromophore than thiocarbonyl, :C:S, or carbonyl itself, :C:O.
(d) Two series of azo dyes of selenium have been prepared and have been shown to possess a marked tinctorial value.
(e) The following new compounds have been prepared:
Since Berzelius published the first resume of the chemistry of selenium, in 1818(1), many articles have appeared in this field. Several reviews(2) of its compounds, including references, have been published, besides the resumes in the chemical dictionaries. These reviews are confined mainly to the inorganic side. No attempt has ever been made to compile a bibliography of selenium organic compounds.
From time to time, articles have appeared, but the field is still a promising one, with many alluring possibilities.
In the perusal of the organic records of the metal, distributed over the span of a century, there are indications of the value of selenium compounds for pharmaceutical and tinctorial uses. An effort has been made to collect these scattered data for critical examination with other analogues and sulphur compounds in particular, and to prepare and study some new organic compounds containing selenium, for the purpose of gaining additional light upon the chemistry of such substances, and in the hope of discovering some which may be of practical service in medicine or elsewhere.
The general conception of selenium is that it is a comparatively rare element. Few realize that it has been known for over a century and that over twenty selenium minerals, containing from one to sixty-six per cent. of the metal, are considered by the mining corporations as important. Beside being a by-product of sulphuric acid manufacture, it is separated also in the electrolytic refining of copper. The demand for the metal is so small that there are half a dozen concerns in the United States either willing to supply gratis any reasonable quantity for research work, or to sell it at cost. In a special report of the National Research Council on selenium(3), it is estimated that there could be produced annually, without making any material additions to the present plants, not less than 300,000 pounds.
In fact selenium has, in recent years, gradually been brought more and more to the attention of the general public through its application to military uses and other purposes. In the glass industry, for example, it was used as decolorizer during the War period. It has been found that it imparts a violet red tint to the pyrex tubing after the latter has been used for a few combustions. The coloration is especially noticeable when a broken piece is examined. This may find an important place in the ceramic industry. In turning off the gas light of the city at day break, in controlling the draft of the factory chimneys, and in regulating the rapidity of the manufacture of sulphuric acid, the selenium cell is an important labor saving factor. In a similar way it is used in automatically lighting and extinguishing light buoys. It also finds application in photometry, wireless telephony, military telegraphy, and army signaling as well as for the transmission of signatures, handwritings, finger prints, and images in general(3)(4).
The question of the vulcanization of rubber also should be considered. Some experiments have been published claiming the similarity of the action of selenium and sulphur on rubber(4)(5). The cost need not be prohibitive, since the supply could be easily increased and the price reduced provided there were a demand. In the personal experience of the writer, when working with the hydrogen selenide gas, the rubber connections of the apparatus soon turned red, and after a few hours were so[Pg 8] clear a red that visitors to the laboratory imagined that the writer was using the ordinary red rubber connections. The rubber thus changed seems to be softer and more elastic than the original; this observation will be followed up.
In this country the National Research Council has created a special committee of seven to investigate the various possible uses of selenium and tellurium.
Duhamel and Rebiere(6)(7) showed that an injection of a trace of red colloidal selenium into rabbits increased urea excretion regularly. In other cases satisfactory results were claimed and the liver showed some lesions. The histological modifications produced by injections into rabbits are most apparent in the liver and kidneys. In the distribution of colloidal preparations in the animal body by injection, Duhamel and Juillard(8) found that the liver contained the greatest amount. Six years later the former(9) used a similar preparation introduced into the animal intravenously, and selenium was again found in the liver, although in smaller quantity.
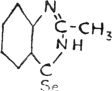
Sulphur compounds have similar physiological action. It is known that triphenylstibine sulphide, or sulphoform, (C6H5)3SbS, has a curative effect in skin diseases, as it liberates “nascent” sulphur on the skin. It is equally natural to expect some organic selenium compound which liberates finely divided selenium to exert a remedial influence on animal bodies. The selenoquinazolone prepared in the course of this research and described more fully in another section of the paper, has this prospect. The quinazolone has the following structure:
Experiments were carried on at L’Institut Pasteur in Paris under the supervision of M. Borel for the treatment of cancer in mice. No human subjects were experimented upon, although results were claimed by using selenides and their oxidized salts.
Selenium dyes were found to be medicinals, although no relation has yet been established between constitution of these organic dyes and their therapeutic value. Wassermann(10) made several eosin preparations, by coupling the sodium derivative with potassium selenocyanide. The red dyestuff thus prepared is stated to be easily soluble in water. Wassermann, Keysser and Wassermann(11) made experiments with it, chemotherapeutically, on animal tumors. When the solution was injected[Pg 9] into mice tumors the latter turned red, accompanied by the softening of the tumor after the third injection and complete resorption after ten injections, unless the dose used was too great for the animal. In that case death often occurred. Good results were also reported, in connection with this experiment, on four different strains of mouse carcinoma and one strain of mouse sarcoma. In the latter case, relief was found sooner but the former disappeared more slowly. Another preparation was made later(12) and introduced into mice intravenously and again found to have good results.
The following is the structure of 2-selenocyanideanthraquinone—
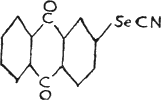
which has also been reported to have medicinal uses(13).
P. Ehrlich and Hugo Bauer(14) synthesized from p.pʹ-diamino-diphenyl-methane the red dye 3,6-diaminoselenopyronine.
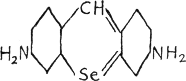
The dye has been used upon mice and caused pronounced edema. The toxicity of both the selenopyronine and the corresponding sulphur compound was compared under similar conditions in the same experiment, and it was found that the selenium dye was toxic in 1/3000 gram, but the sulphur dye was toxic in 1/2500 gram per twenty gram weight of the animal.
This physiological activity was noted years ago with the inorganic compounds of selenium and Berzelius(15) described the poisonous effect of hydrogen selenide quite impressively; “In order to get acquainted with the smell of this gas I allowed a bubble not larger than a pea to pass into my nostril; in consequence of its smell I so completely lost my sense of smell for several hours that I could not distinguish the odor of strong ammonia even when held under my nose. My sense of smell returned after five or six hours, but severe irritation of the mucous membrane set in and persisted for a fortnight.” The writer has been working on the gas for some time and was also quite seriously affected once, the injury persisting for many[Pg 10] days. That it is more poisonous than the hydrogen sulphide is well known.
Bruere(16) showed that when hydrogen sulphide was passed into blood solution sulphemoglobin was produced in considerable quantity, due to the chemical action of sulphur and hematin. He stated further that sulphemoglobin may be found in animal blood when a large amount of the gas has been inhaled. He made selenhemoglobin in the same manner. Sixteen years later, Clarke and Hurtley also proved that selenhemoglobin may be made by passing hydrogen selenide into blood(17). These experiments may be interpreted to mean that the oxy-hemoglobin is transformed into an organic complex of sulphur or selenium, and that the transference may be more rapid and powerful in the case of hydrogen selenide.
Biological investigations have sufficiently proved that dyestuffs of the phenazine, oxazine, thiazine, acridine series show an injurious effect on protozoa, especially those dyes containing substituted amino groupings and of a simple structure(18). In the case of the thiazine dyes of the methylene blue class, the physiological importance has well recognized in their use as feeble antiseptics and analgesics. Ehrlich and Guttmann(19) initiated the use of methylene blue as an antiperiodic and its use in that line has been continued.
In the field of the selenazine dyes, pharmacologists have not yet paid much attention to them, on account of the newness of the discovery, but P. Karrer claims that they are indisputably “vital dyestuffs”(20). The prospect of synthesizing selenazine dyes and their use as drugs seems to be bright, judging from the fact that they are easily prepared and capable of many combinations, especially of the ease with which they form organic complexes with arsenic compounds.
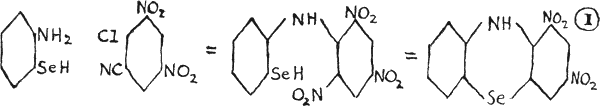
Formula (I) is known, as 1, 3-dinitrobenzoselenazine(21), which was obtained by the action of picryl chloride on the zinc salt of o-aminoselenophenol; the product (picrylaminoselenophenol) being then treated with alkali and thus converted to the dye, which upon experimentation showed marked effects upon protozoa and bacteria.
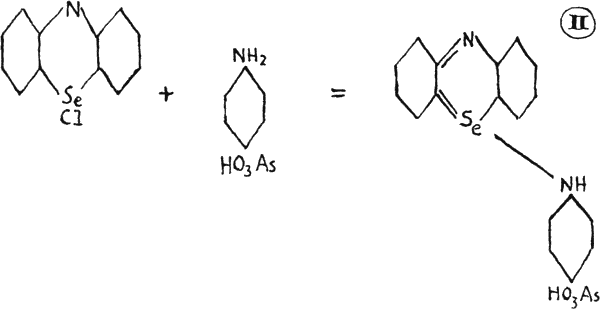
Formula (II), known as 3-(p-phenylarsonic)-aminoselenazine, is red in dilute alkali and green in mineral acid, and is a typical dye in a series from the coupling of selenodiphenylamine with arsenic compounds. All possess similar toxicity as the thiazine dyes(20). Other selenazines are listed in the bibliography(22).
No less than half dozen thioureas are commonly used as drugs. Thiourea itself paralyzes the nerve centers, and is employed commercially for photograph fixing and for removing stains from negatives; thiuret, C6H7N3S2, serves as a substitute for iodoform; thiosinamine-ethyliodide, or tiodine, IH5C2H2NCSNHC3H5, is used for relief of lesions of the central nervous system; allylthiourea or thiosinamine, (NH2)SC.NHCH2CH:CH2, for aiding the absorption of connective tissues, for treatment of burns, keloids, urethral diseases, sclerotic conditions of the ear(23).
Selenocarbamide and a number of its derivatives have been prepared and studied. One class of seleno ureas has been patented as pharmaceutical products by Chem. Fabrik von Heyden(24), and are prepared by the action of hydrogen selenide on alkylcyanamides,
RNH.CHN + H2Se = RNH.CSe.NH2
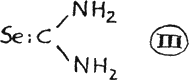
They possess pronounced therapeutic value and, serve as intermediate products in the production of more stable alkyl halide additive compounds. Other carbamides ranging from seleno urea itself(25), (III) and a cyclic urea(26) (IV) are described in the literature:
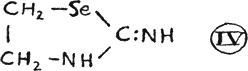
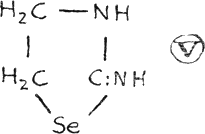
The latter, known as ethylene-selenourea, may be classified also in the azole group as 2-iminotetrahydroselenazole (V).
The literature for the other normal carbamides is listed in the bibliography(27).
Selenoantipyrines, selenosaccharine, selenoindigoes have also been prepared.
Thiophene and its derivatives are of considerable therapeutic interest. Thiophene itself is found to be useful in lessening the elimination of sulphuric acid in urine, and is employed in the dermatological practice. Sodium thiophene sulphonate, thiophenetetra-bromide, thiophene diiodide, are all medicinals(23).
A number of selenophenes are recorded in the literature. Their relation to the selenazoles may be easily seen from the following formulas:
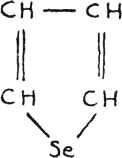
Selenophene
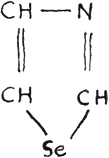
Selenazole
Dimethyl selenophene was prepared from acetonyl acetone and phosphorous pentaselenide,
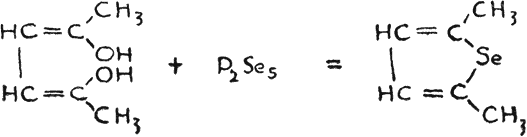
The compound thus obtained is stated to have the same odor as thiophene, but no mention is made in regard to its uses(28). Selenophene was prepared from sodium succinate and phosphorous triselenide, or by conducting ethylselenide through hot tubes(29).
Some selenazoles find application also in medicine. At present only the isoazoles are known to have physiological uses. One of them was prepared from anthraquinone selenocyanide, by the action of ammonia under pressure(30).
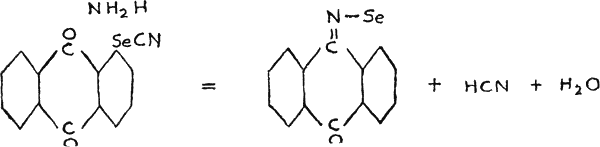
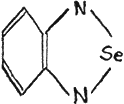
Another type of azoles, benzoselendiazole (piaselenol) and five of its derivatives, have been also described as medicinals(31). The diazole itself has the following structure,
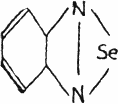 or
or
Diazoles of the following structure are also known, but no data were found, regarding their physiological action(32):
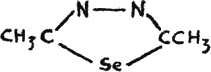
Dimethyl-seleno-diazole
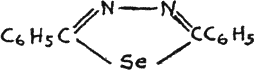
Diphenyl-seleno-diazole
Sulphides and disulphides have curative power. Dimethylsulphide is used for internal treatment, di-o-aminophenyldisulphide is used for intramuscular injections. Diallyl sulphide is also a medicament. Methyl selenide has some effect on the internal parts of the body(33). Hanzlik and Tarr(34) at the American University Experimental Station, showed that a number of selenium compounds act as skin irritants: e.g., dichlorodiethyl selenide, dichlorovinyl selenide, trichlorodiethyl selenide and selenium mustard oil. The first mentioned proved as potent as the sulphide, but the others fell somewhat below in their effects. Diantipyryl selenide is another therapeutical agent(35).
The diselenides occupy an important place of their own. The selenophenols do not remain unchanged in the air, but are always oxidized to the diselenides, which can be again reduced to the selenophenols. So far only the diselenides of anthraquinone and their phenols are recognized remedies(36).
Many of the seleno organic compounds are colored, while the corresponding sulphur derivatives are colorless.
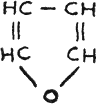
Furane, colorless
liquid
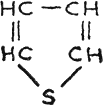
Thiophene, colorless
liquid
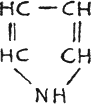
Pyrrol, colorless liq.
but turns brownish in air
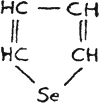
Selenophene, yellow liq.
after repeated extraction[B]
This brings selenophene more akin to pyrrole than thiophene, but the group -NH- in the molecule of pyrrole is an auxochrome. The selenium atom in a cyclic compound also acts like an auxochrome.
Selenoantipyrine(37),
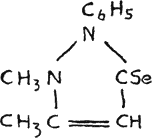
forms pure yellow crystals from alcohol, while the corresponding compounds of oxygen and sulphur are colorless.
Similarly, the 2-methyl-4-selenoquinazolone is deep brown in color, while the thio compound, prepared by Bogert and Hand(38) is light brown or yellow and the corresponding oxygen compound is colorless or nearly so.
Diethyl selenide (C2H5)2Se, is a yellowish heavy oil of unpleasant odor. It combines readily with chlorine to form a chloride (C2H5)2SeCl2, and the latter is oxidized by nitric acid to form an oxide (C2H5)2SeO,(39). Diethyl sulphide is a colorless syrupy liquid, as well as diethyl amine and diethyl ether.
The gradation of color is quite pronounced in the case of selenonaphthene quinone(40).
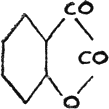
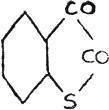
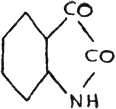
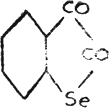
It would be most natural to conclude that the chromophore :CS is more powerful than :CO, and that :CSe is most powerful of all, as shown in our study of quinazoline compounds. It would equally follow that :S is a more powerful color-forming radical in a cyclic compound than that of :O; and :NH than that of :S; and :Se again most powerful of the whole series.
Lesser and Weiss(41) in their research on selenoindigo stated that the selenium dyestuff, on account of its greater molecular weight than sulphur, shows a deeper blue. This hypothesis meets a difficulty in the case of coumorandione, thionaphthenequinone and isatin series, where the -NH- radical has an atomic weight of 15, and -S- 32, and showed the reversed order of color. This seems to be the case in the selenophene series also. Therefore this theory is not without exceptions.
The diselenides present a very interesting study also. Methyl disulphide is colorless, but methyldiselenide(42) is a reddish yellow liquid. Methyl disulphide only becomes yellow when it is treated with chlorine, and in such cases (CH3)2S2Cl2 is formed(43), in yellow rhombic crystals. Ethyldisulphide is colorless: ethyldisulphidedichloride is a faint yellow oil(44). But the corresponding ethyldiselenide is a red liquid(45). Phenyl disulphide is colorless, and phenyldisulphide dibromide is of mother-of-pearl appearance, and practically colorless(46), while phenyl diselenide forms pure yellow needles(47), and phenyldiselenide dibromide orange red ones.
While phenyldisulphide is colorless, when an auxochrome group is added, such as NH2, the compound is colored. This is the case with o-diaminodiphenyldisulphide(48) which is yellow both in solution and in crystalline form. In other words, an auxochrome in addition to the chromophore group transforms a colorless chromogen into a colored one. Therefore groups like -S.S- and -Se.Se- are chromophores in the same sense as -N:N-. This is in agreement with the chromophore ideas of Hugo Kaufmann. The -Se.Se- is a more powerful chromophore than -S.S-.
This brings one directly to the inquiry as to why 2-phenylbenzoselenazole, which contains a :Se radical, should be colorless; and that even 6-nitro-2-phenylselenazole, with the addition of a chromophore NO2, should be only faintly colored. The benzothiazoles, their isomers and derivatives are mostly colorless, and similar causes are probably responsible in the case of the phenylbenzoselenazole, for its lack of color. But when this[Pg 16] selenazole is combined with another chromophore, for example an azomethine grouping, the result is a more positively colored compound (in this case benzalaminoselenazole), the crystals being yellow. The corresponding thiazole derivative is light colored.
The tinctorial value of the selenium derivative is further evidenced by the ease with which it forms azo dyes and the deep colors of the latter. This was observed when 6-amino-2-phenylbenzoselenazole was diazotized and coupled with B-naphthol, salicylic acid, etc. The corresponding aminothiazole has been considered difficult to diazotize, on account of its insolubility in hydrochloric acid, cold or hot, but the aminoselenazole dissolves readily and completely, the coupling is almost instantaneous, and the dyes obtained are mostly red and of metallic lustre. In view of the stability of benzoselenazoles toward hot concentrated acids (with the exception of nitric, when nitration ensues) and alkalis, these dyes may prove of some commercial interest.
The azole dyes of the benzoselenazole have been exposed to light for weeks, and also exposed to acids and alkalis, and have been found to be quite fast.
Busch prepared quinazolines by the action of o-amino or o-nitro benzylamine with phosgene, and thioquinazolines with carbon disulphide(51):
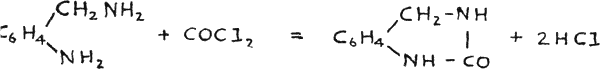
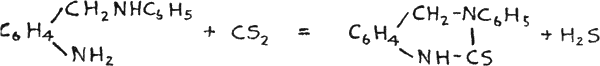
Accordingly the same reaction was tried with o-nitrobenzylamine, prepared by the method of Lellmann and Stickel(50), using carbon diselenide(51). The reaction seemed to work, but the mixture formed was difficult to extract and it appeared that other reactions took place at the same time, due to the impurity of the carbon diselenide, as the latter has never been prepared in the pure state.
Another method, which is equally attractive because of its simplicity, is that of Gabriel and Stelzner(52),
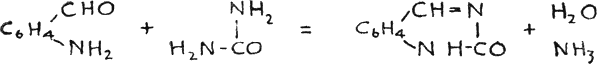
In accordance with the above reaction o-aminobenzaldehyde should work with equal ease with selenocarbamide, but the initial materials were not available.
The reaction which was used successfully was that of Bogert, Breneman and Hand(53),
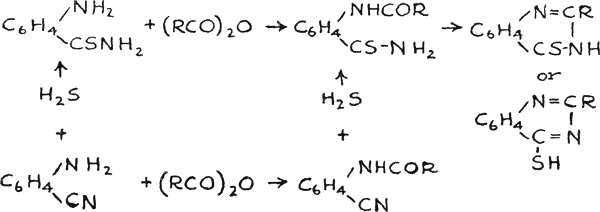
The hydrogen selenide used in the reaction was prepared from FeSe by the action of hydrochloric acid, or by heating paraffin and selenium, in the proportion of four to one respectively, at 335° to 350°C(54).
The selenoquinazoline was prepared from anthranilic nitrile by the following methods—the anthranilic nitrile being prepared from o-nitraniline(57),
(a) 20 grams of acetyl-anthranilic nitrile was dissolved in absolute alcohol, and dry hydrogen selenide and dry ammonia passed into the solution for three hours. The quinazoline crystallized out gradually on cooling was filtered out and recrystallized from dilute alcohol. The yield was about ten per cent.
(b) 10 grams of acetyl-anthranilic nitrile was heated in a sealed tube at 110° with alcohol saturated at zero degrees with dry hydrogen selenide and dry ammonia. After five hours, the tube was taken out and the quinazoline crystallized out on cooling. Yield was about sixteen per cent.
As hydrogen selenide was somewhat unstable and did not dissolve freely in alcohol, freshly prepared sodium selenide was used in the following method and was found to be more satisfactory. It was prepared from sodium hydroxide in absolute alcohol by passing dry hydrogen selenide into the solution for about three hours. In the beginning and end of the reaction, nitrogen was used to exclude the oxygen of the air. The selenide was collected and dried in an atmosphere of nitrogen, and then in a vacuum, in presence of phosphorus pentoxide. When thus prepared, sodium selenide was colorless, but on exposure to[Pg 18] air it turns reddish and finally dark colored. The C. P. selenide on the market was black and was found to be entirely useless.
(c) 20 grams of anthranilic nitrile and fifty grams of sodium selenide were mixed and heated in a distilling flask in an atmosphere of nitrogen, and forty grams of acetic anhydride dropped into the flask very slowly. The temperature was kept at 115° for half an hour and then raised to distill off the acetic acid formed in the reaction, as the condensation hardly went to completion in the presence of any trace of acetic acid. The whole process took an hour and a half. The flask was removed from the oil bath and, after cooling, dilute alkali was run in, in successive portions, to dissolve out the quinazoline. Into the clear alkaline extracts carbon dioxide was bubbled for an hour, and common salt then added. The precipitate was recrystallized several times from twenty-five per cent. alcohol. The yield was from twenty to twenty-five per cent.
(d) 10 grams of anthranilic nitrile, twenty grams of acetic anhydride, and twenty-five grams of sodium selenide were mixed in a sealed tube and heated together for three hours and a half at 110°-115°. The condensation product was crystalline when the tube was cooled to room temperature. The contents of the tube were extracted with dilute alkali as before, filtered, precipitated by carbon dioxide, and recrystallized from dilute alcohol. The yield was not over twenty per cent.
(e) An attempt was made to make o-aminobenzoselenamide, and from the latter, by treatment with acetic anhydride, to form the quinazoline, but the yield of the amide was too small to carry the reaction further.
The substance prepared by the above methods crystallizes from dilute alcohol in needles or prisms of dark brown color. It melts at 213.5° (corr.). It dissolves readily in hot alcohol but on concentration sometimes forms a sticky mass with a peculiar but not unpleasant odor. It dissolves readily in alkalis and is slightly soluble in hot benzene and chloroform, but insoluble in hot water. Crystals purified from (a) were analyzed and gave the following results:
| Calculated for C9H8N2Se |
Found | ||
|---|---|---|---|
| I | II | ||
| Carbon | 48.38% | 48.45% | 48.62% |
| Hydrogen | 3.61 | 3.82 | 3.52 |
| Nitrogen | 12.55 | 12.51 | 12.66 |
| Selenium | 35.46 | 35.60 | 35.42 |
The crystals on standing in the presence of air and light decomposed with separation of finely divided selenium and methyl quinazolone.
In the quantitative determination of selenium in quinazoline[Pg 19] the method adapted by Becker and Meyer was found to be quite satisfactory(56). Other methods are listed in the bibliography(57).
In the ultimate analysis of carbon and hydrogen, the ordinary absolute method was followed with the use of copper oxide, lead chromate, and lead peroxide in the tube. In the determination of nitrogen the ordinary absolute method was also followed excepting that a considerable quantity of specially prepared lead chromate powder was mixed with the sample in a number six porcelain boat. This was found to be desirable when the dinitroselenazole was burned. Selenium dioxide, which is a solid, seems to be formed in the tube and carried away by the current of carbon dioxide with some difficulty. In such a case the analysis usually took four hours after the combustion had actually started. In the carbon and hydrogen determination selenium dioxide was easily absorbed in the presence of oxygen gas.
The first method employed was a modification of the method described by Fromm and Martin(58). Method (b) is an adaptation of the method for preparing benzothiazoles.
(a) Twenty grams of benzanilide was mixed in a pyrex flask with 160 grams of selenium dust and the flask placed in a nitrate bath under an air condenser. After heating for an hour at 220°C., the temperature was raised to 250°C., and kept at 250°-280°C. for sixteen hours. The dark mass was extracted with hot concentrated HCl, the acid extracts filtered through glass wool using a hot water funnel. The combined extracts were poured into a large volume of water when the selenozole precipitated out immediately; it was recrystallized from alcohol. In some cases it was necessary to dissolve in hot HCl again and to recrystallize. The yield was twelve per cent.
The above method has the disadvantage that water is formed in the reaction and this in turn reacts upon benzanilid at the higher temperature necessary (as selenium only melts at 217°C.), decomposing the benzanilid into aniline and benzoic acid.

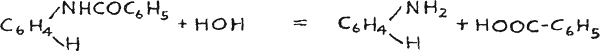
Furthermore benzanilid boils at 160°C. and at such high temperatures as 250°C. and over some of it is apt to be driven off.
(b) 106 grams of benzaldehyde were heated with 93 grams of redistilled aniline at 120°C., for two hours or until the solution was clear. The clear benzalaniline was then poured into 160 grams of selenium dust in a pyrex flask on a sand bath, the flask being connected with an air condenser as before. In order to distribute the flame to better advantage over the bath, an air space was made between the Meker burner and the bath by introducing a wire gauze. Hydrogen selenide was evolved freely. Complete reaction took three days. The extraction and recrystallization were the same as in the former case. The yield was sixty per cent.
The selenazole crystallizes in colorless long needles, melting at 117.5°C. (corr.) Fromm and Martin(58) gave the melting point as 117°C. It is insoluble in water, and in the following solvents it is lightly soluble in the cold, more easily hot: ether, methyl alcohol, acetone, acetic acid, acetic anhydride, chloroform, and nitrobenzene. It is difficultly soluble in ethyl alcohol, ethyl acetate, and carbon tetrachloride, in the cold, but easily soluble hot.
The mononitro derivative of the selenazole, 6-nitro-2-phenylbenzoselenazole, was prepared by nitration with nitric acid at a low temperature:
Twenty-five grams of the selenazole were dissolved in 150 grams of concentrated sulphuric acid, keeping the temperature below the room temperature until complete solution took place. It was then cooled on a freezing mixture and a mixture of sulphuric and nitric acids (previously prepared and cooled by mixing 9.5 grams of nitric and fifteen grams of sulphuric acids) slowly dropped into it in the course of half an hour, using mechanical stirring for four hours. The solution was then poured into two liters of water (ice water), filtered, dried, and recrystallized from acetic acid, and alcohol with the help of animal charcoal. The yield was 95 per cent.
This nitro compound crystallizes in flattened needles of a light yellow color. It melts at 202.4°C. (corr.). It is very insoluble in water; but soluble in hot acetic acid, acetic anhydride, nitrobenzene, nitrotoluene, toluene, benzene, alcohol, and difficultly soluble when cold. The crystals were analyzed and gave the following results,
| Calculated for C13H8N2O2Se |
Found | ||
|---|---|---|---|
| I | II | ||
| Nitrogen | 9.24% | 9.36% | 9.48% |
The conversion of mononitro compound to 6-amino-2-phenylbenzoselenazole was accomplished by the action of tin and hydrochloric acid as follows:
30.3 grams of nitro compound were mixed with 42 grams of twenty mesh tin in a liter flask, immersing the latter in cold water. 175 cc. of conc. HCl were slowly added to the flask. In some cases it was necessary to apply initial heating but when once the reaction started it took place rapidly. After the effervescence had abated, the flask was heated over a free flame, under a return condenser, for two hours. The solution usually turned to a pasty mass, due to the formation of a tin double salt. The mixture was dissolved in a large volume of water and heated on a water-bath, the precipitate filtered out, washed, and preserved. The clear filtrate was treated with concentrated alkali, in excess, the separated amine collected, washed with water, dried and recrystallized from alcohol, using bone-black. The precipitate set aside was treated with strong alkali, the insoluble residue washed, recrystallized, and added to the main product. The yield was 75 per cent.
This amine crystallizes from alcohol in fine yellowish needles, melting at 201.2°-202.3°C (corr.). It is insoluble in water and ether, difficultly soluble in the hot; and fairly soluble in aniline. A pure sample was analyzed and gave the following results:
| Calculated for C13H10N2Se |
Found | ||
|---|---|---|---|
| I | II | ||
| Nitrogen | 10.25% | 10.34% | 10.42% |
| Carbon | 57.18 | 57.17 | 57.00 |
| Hydrogen | 3.69 | 3.79 | 3.85 |
Five grams of the monoamino compound were mixed with powdered KOH, heated together until the mixture just melted, and maintained in that state for a few minutes. When the latter had cooled down to room temperature, cold water was poured over the mixture. The filtered solution was acidified until no further precipitate was formed. The precipitate was collected and recrystallized from water, m.p. 121°C.
One gram of this solid was placed in a test tube, provided with a cork and a delivery tube, and heated with soda lime; a liquid with the smell of benzene was collected in another test tube cooled with water. When this liquid was treated with a few drops of nitric acid mixture the smell of nitrobenzene was given off. A gram of the crystals was heated with concentrated sulphuric acid and alcohol when the odor of ethyl benzoate was noted.
Five grams of the monoamino selenazole were heated on a water-bath with 10 cc. of acetic anhydride until the solution was clear, which took about two hours. 100 cc. of water were poured into the mixture, which was then neutralized with dilute am[Pg 22]monium hydroxide. The precipitate was filtered, decolorized by animal charcoal, and recrystallized from dilute alcohol.
The acetyl compound, 6-acetamino-2-phenylbenzoselenazole, forms colorless crystals, melting at 188.1°-.7°C. (corr.). It is insoluble in ether, benzene, carbon disulphide; slightly soluble in toluene; soluble in alcohol, ethyl acetate, amyl acetate, acetone, and acetic acid. A pure sample was analyzed and gave the following result,
| Calculated for C15H12N2SeO |
Found | ||
|---|---|---|---|
| Nitrogen | 8.88% | 8.92% | 8.68% |
Five grams of the monoamino compound were dissolved in 200 cc. absolute alcohol with the addition of 3 cc. of benzaldehyde and the clear solution was boiled on a water-bath, with a return condenser, for two hours. After the solution was boneblacked, the yellow precipitate was recrystallized from carbon disulphide. The yield was 90 per cent.
It crystallizes in yellow plates, melting at 156.7°-157.6°C., soluble in benzene, ether, ethyl alcohol, carbontetrachloride, acetone, but difficultly soluble in ligroin. An analysis of the crystals showed the following result,
| Calculated for C20H14N2Se |
Found | ||
|---|---|---|---|
| Nitrogen | 7.75% | 7.92% | 7.68% |
Five and four tenth grams of the monoamino compound were dissolved in hot conc. HCl, cooled in ice, and diazotized with sodium nitrite solution, until starch iodide paper showed excess nitrous acid. The diazotization was performed in ice, with mechanical stirring, and required about an hour. The diazo solution was poured into a solution of 3 grams B-naphthol in 8 grams of NaOH and 60 cc. of water, while gradually stirring. A very deep red solution formed. This was acidified with excess HCl, salted out by NaCl, and crystallized from aniline-alcohol mixture. In the pure state, it is a deep red powder, with a metallic lustre when rubbed, melting at 284.2°C. An analysis showed the following result,
| Calculated for C23H15N3OSe |
Found | |
|---|---|---|
| Nitrogen | 9.81% | 9.75% |
The nitration for the production of dinitro derivative was at first carried out under the same conditions as in the preparation[Pg 23] of mononitro compound and after the latter was formed more nitric acid mixture was added with the addition of heat: conc. sulphuric acid, keeping it below room temperature. It was then cooled in a freezing mixture and half the volume of a nitric acid mixture (prepared and cooled by mixing 19 grams of nitric and 30 grams of sulphuric acids) was introduced very slowly to the selenazole solution through a dropping funnel, maintaining at this temperature for two hours (using mechanical stirring). The remaining half of the nitric acid mixture was then slowly introduced and the flask was heated on a water-bath for two hours. The solution was poured into two liters of water, the precipitate filtered off, dried and recrystallized several times from acetic acid. The yield was 80 per cent.
This dinitro compound crystallizes in fine yellow needles, m. p., 246.8°C. (corr.), very insoluble in water, but soluble in hot acetic acid, acetic anhydride, nitrobenzene, nitrotoluene, ethyl alcohol, and difficultly soluble cold. It was analyzed and the following results were found,
| Calculated for C13H7N3O4Se |
Found | ||
|---|---|---|---|
| I | II | ||
| Nitrogen | 12.07% | 12.30% | 12.12% |
The conversion of the dinitro to diamino derivative was accomplished in the same manner as the reduction of the mononitro derivative excepting that twice as much tin and HCl were used.
This diamino compound crystallizes in yellowish glistening needles from alcohol and pyridine; m. p., 269°-270.5°C.; was analyzed and gave the following results,
| Calculated for C13H11N3Se |
Found | ||
|---|---|---|---|
| I | II | ||
| Nitrogen | 14.6% | 14.4 | 14.7 |
The diacetyl and dibenzylidene compounds were also prepared from these diamino derivatives. The former crystallizes in cubes from dilute alcohol; m. p., 307°C (Corr.) and the latter in beautiful yellow plates from carbon disulphide, m. p., 195°-196°C. (Corr.). An analysis of these two compounds showed the following results,
| Calculated for C17H15N3O2Se |
Calculated for C27H19N3Se |
Found | ||
|---|---|---|---|---|
| I | II | |||
| Nitrogen | 9.05% | 11.21% | 9.21% | 11.43% |
Both the monoamino and the diamino derivatives form intensely colored dyes when diazotized and coupled with phenols and aromatic amines. The dyes formed are fast to light. In the following table silk is given to represent the fabrics used. Wool and cotton were dyed similar shades, though with slight variation. Each silk sample was dyed in acid or alkaline baths as indicated and each bath contained 0.01 gram in twenty cc. solution:
| Coupler | Diazo. monoamine |
On silk |
Diazo. diamine |
On silk |
|---|---|---|---|---|
| Phenol | deep red (alk) |
v. light yellow |
(alk) deep red |
yellow |
| Dimethylaniline | orange (acid) |
light yellow |
orange-red (acid) |
yellow |
| P-nitraniline | light brown (acid) |
brownish (acid) |
grayish brown | |
| P-toluidine | light brown (acid) |
brownish (acid) |
brownish | |
| Pyrogallic acid |
dark brown (acid) |
grayish | dark brown (alk) |
grayish |
| Salicylic acid |
reddish (alk) |
light brown | red (alk) |
brown |
| B-naphthol | deep red (alk) |
pink | deep red (alk) |
red |
| Sulphanilic acid |
light brown (alk) |
brownish | brown (alk) |
brown |
| A-naphthylamine | light brown (acid) |
brownish | yellow (acid) |
yellow |
| Resorcinol | purple (alk) |
red | dark purple (alk) |
deep red |
Yü-Gwan Chen was born in Nanking, China, March 8, 1893. After graduation from college in 1915, he further studied Chinese classics, 1915-16. He entered Case School of Applied Science, Cleveland, Ohio, as a special student in the Department of Chemistry, 1916-17. He registered at Columbia University to pursue graduate work in chemistry under the Faculty of Pure Science; and was awarded the degree of Master of Arts in 1918. From September 1919 to June 1922, he has been pursuing research in organic chemistry in the research laboratories of Havemeyer Hall, Columbia University.
[A] Not a new compound but prepared by a different method.
[B] Note—dimethyl selenophene, however, is colorless.
The diagram of 3-(p-phenylarsonic)-aminoselenazine, labelled (II), has been adjusted to make its structure clearer.
The "l" in "2HCl" in the first equation under "Preparation of 2-methyl-4-selenoquinazolone" was missing in the original.
The following is a list of changes made to the original. The first line is the original line, the second the corrected one.
selnoquinazolone prepared in the course of this research
selenoquinazolone prepared in the course of this research
on four different strains of mouse carcenoma and one strain
on four different strains of mouse carcinoma and one strain
in consequence of its smell I so completely loss my sense of smell
in consequence of its smell I so completely lost my sense of smell
of the phenazine, oxasine, thiazine, acridine series show
of the phenazine, oxazine, thiazine, acridine series show
and Se again most powerful of the whole series.
and :Se again most powerful of the whole series.
simplicity, is that of Babriel and Stelzner(52),
simplicity, is that of Gabriel and Stelzner(52),
on cooling was filtered out and recrystalized from dilute alcohol.
on cooling was filtered out and recrystallized from dilute alcohol.
a sealed tube at 110° with alcohol saturated at zero degree with
a sealed tube at 110° with alcohol saturated at zero degrees with
It was prepared from Sodium hydroxide in absolute alcohol
It was prepared from sodium hydroxide in absolute alcohol
took an hour and half. The flask was removed from the oil
took an hour and a half. The flask was removed from the oil
The precipitrate was recrystallized several times
The precipitate was recrystallized several times
(e) An attempt was made to make o-aminobenzselenamide,
(e) An attempt was made to make o-aminobenzoselenamide,
It dissolves readily in alkalies and is slightly soluble
It dissolves readily in alkalis and is slightly soluble
This was found to be desirable when the dinitroselezazole was burned.
This was found to be desirable when the dinitroselenazole was burned.
into a large volume of water when the selenozole precipitated out
into a large volume of water when the selenazole precipitated out
it was necessary to dissolve in hot HCl again and to recrystalize.
it was necessary to dissolve in hot HCl again and to recrystallize.
(b) 106 grams of benaldehyde were heated with 93 grams of
(b) 106 grams of benzaldehyde were heated with 93 grams of
It is difficulty soluble in ethyl alcohol, ethyl acetate, and
It is difficultly soluble in ethyl alcohol, ethyl acetate, and
toluene, benzene, alcohol, and difficulty soluble when cold.
toluene, benzene, alcohol, and difficultly soluble when cold.
from alcohol in fine yellowish needles, melting at 201.2°202.3°C
from alcohol in fine yellowish needles, melting at 201.2°-202.3°C
carbontetrachloride, acetone, but difficulty soluble in ligroin.
carbontetrachloride, acetone, but difficultly soluble in ligroin.
19 grams of nitric and 30 grams of sulphuric acids was introduced
19 grams of nitric and 30 grams of sulphuric acids) was introduced
in the same manner as the reduction of mononitro derivative
in the same manner as the reduction of the mononitro derivative
This diamino compound crystalizes in yellowish glistening needles
This diamino compound crystallizes in yellowish glistening needles
compounds were also prepared from this diamino derivatives.
compounds were also prepared from these diamino derivatives.
The former crystalizes in cubes from dilute alcohol;
The former crystallizes in cubes from dilute alcohol;