This eBook is for the use of anyone anywhere at no cost and with almost no restrictions whatsoever. You may copy it, give it away or re-use it under the terms of the Project Gutenberg License included with this eBook or online at www.gutenberg.org
Title: A Textbook of Assaying: For the Use of Those Connected with Mines.
Author: Cornelius Beringer and John Jacob Beringer
Release Date: July 3, 2006 [eBook #18751]
Language: English
Character set encoding: ISO-8859-1
***START OF THE PROJECT GUTENBERG EBOOK A TEXTBOOK OF ASSAYING: FOR THE USE OF THOSE CONNECTED WITH MINES.***
Transcriber's Note:
Parentheses have been added to clarify fractions. Letters in brackets with a = sign before it means that the letters have a macron over them, e.g. H[=A=c] signifies that the Ac has a macron over it.
Minor typographical errors have been corrected. Footnotes have been moved to the end of the chapter, and all advertisements have been moved to the end of the book.
ASSOC. OF THE ROYAL SCHOOL OF MINES; FELLOW OF THE CHEMICAL SOCIETY AND OF THE INST. OF CHEMISTRY; PRINCIPAL OF THE CAMBORNE MINING SCHOOL; AND LATE PUBLIC ANALYST FOR THE COUNTY OF CORNWALL.
LONDON:
CHARLES GRIFFIN AND COMPANY, Limited,
EXETER STREET, STRAND.
1904.
The continued popularity of the present work, the last edition of which was published only a little over a year ago, continues to be a source of gratification to the publishers, who have much pleasure in issuing the present edition.
January 1904.
The principal changes in this edition are additions to the articles on Gold, Cyanides, and Nickel, and a much enlarged Index. The additional matter covers more than forty pages.
J. J. BERINGER.
Camborne,
January 1900.
The Text-book now offered to the public has been prepared to meet the existing want of a practical "handy book" for the Assayer.
To mining men the word "assaying" conveys a sufficiently clear meaning, but it is difficult to define. Some writers limit it to the determination of silver and gold, and others imagine that it has only to do with "furnace-work." These limitations are not recognised in practice. In fact, assaying is becoming wider in its scope, and the distinction between "assayers" and "analysts" will in time be difficult to detect. We have endeavoured rather to give what will be of use to the assayer than to cover the ground within the limits of a faulty definition.
At first our intention was to supply a description of those substances only which have a commercial value, but on consideration we have added short accounts of the rarer elements, since they are frequently met with, and occasionally affect the accuracy of an assay.
Under the more important methods we have given the results of a series of experiments showing the effect of varying conditions[Pg vii] on the accuracy of the process. Such experiments are often made by assayers, but seldom recorded. Statements like those generally made—that "this or that substance interferes"—are insufficient. It is necessary to know under what conditions and to what extent.
Students learning any particular process cannot do better than repeat such a series of experiments. By this means they will, at the same time, acquire the skill necessary for performing an assay and a confidence in their results based upon work under different conditions.
The electrolytic method of copper assaying given under Copper is a modification of Luckow's; it was introduced by us into the offices of the Rio Tinto Copper Company, and has been in use for many years with success. This modification is now employed in copper-works in Spain, Germany, and England, and is used in place of the dry assay for the commercial valuation of copper ores.
We have adhered to the gram and the "c.c." as the units of weight and volume. Those who prefer working with grains and grain-measures can use the figures given, multiplied by ten. For example:—When 1 gram is mentioned, 10 grains should be used, and 10 grain-measures take the place of 1 "c.c." It is not advisable to mix the two systems, as by using gram weights and grain-measures.
We have intentionally to a large extent omitted to mention the names of those who have originated or modified the various processes. The practice of naming a process after its discoverer has developed of late years, and is becoming objectionable. It is a graceful thing to name a gas-burner after Bunsen, or a condenser after Liebig; but when the practice has developed[Pg viii] so far that one is directed to "Finkenerise" a residue, or to use the "Reichert-Meissl-Wollny" process, it is time to stop.
We are indebted to the standard works of Allen, Crookes, Fresenius, Lunge, Michell, Percy, and Sutton, and wish to express our sense of special indebtedness to Mr. Richard Smith, of the Royal School of Mines. One or two of the illustrations are taken from Mr. Sexton's excellent little book on Qualitative Analysis. Our obligation to some others is mentioned in the text.
Finally, we have to thank for assistance in the experimental work Messrs. Bailey, Beswick, Clarke, Grant, Higgins, and Smith.
THE AUTHORS.
Camborne, Nov. 1889.
| PART I. | |
| CHAPTER I. | |
| INTRODUCTORY. | |
| Page | |
| Object of assaying | 1 |
| Sampling | 1 |
| Drying: determination of moisture | 5 |
| Calculation and statement of results | 7 |
| Laboratory books and report forms | 9 |
| Quantity to be taken for an assay | 11 |
| Exercises | 14 |
| CHAPTER II. | |
| METHODS OF ASSAYING.—DRY GRAVIMETRIC METHODS. | |
| Methods of assaying | 15 |
| Gravimetric methods | 15 |
| Mechanical separations | 16 |
| Dry assays | 16 |
| (a) Fluxes | 16 |
| (b) Reducing agents | 21 |
| (c) Oxidising agents | 22 |
| (d) Apparatus | 24 |
| CHAPTER III. | |
| WET GRAVIMETRIC METHODS. | |
| Wet gravimetric methods | 27 |
| (a) Solution | 29 |
| (b) Precipitation | 30 |
| (c) Filtration | 31 |
| (d) Drying and igniting | 32 |
| CHAPTER IV. | |
| VOLUMETRIC ASSAYS. | |
| Titrometric assays | 35 |
| (a) Standard solutions | 36 |
| (b) Standardising | 37 |
| (c) Methods of working | 42 |
| (d) Indirect titration | 43 |
| Colorimetric assays | 44 |
| Gasometric assays | 44 |
| CHAPTER V. | |
| WEIGHING AND MEASURING. | |
| Weighing | 47 |
| Measuring liquids | 49 |
| (a) Graduated flasks | 49 |
| (b) Pipettes | 50 |
| (c) Burettes | 51 |
| Measuring gases | 52 |
| CHAPTER VI. | |
| REAGENTS. | |
| Acids, &c. | 54 |
| Bases, salts, &c. | 59 |
| CHAPTER VII. | |
| Formulæ, equations, &c. | 68 |
| CHAPTER VIII. | |
| SPECIFIC GRAVITY. | |
| Introductory | 75 |
| Determination of specific gravity— | |
| (a) Hydrometers | 76 |
| (b) Specific gravity bottles | 78 |
| Calculations depending on specific gravity | 84 |
| PART II. | |
| CHAPTER IX. | |
| SILVER, GOLD, PLATINUM, CYANIDES, MERCURY. | |
| Silver—Detection | 87 |
| Dry assay | 87 |
| (1) Scorification | 88 |
| (2) Pot assays, average ores | 90 |
| " ores with metallic oxides | 91 |
| " ores with metallic sulphides | 91 |
| Explanatory notes on the fusion | 93 |
| The effect of charcoal, flour, &c. | 94 |
| The effect of nitre | 95 |
| The effect of mineral sulphides | 95 |
| (3) Cupellation | 98 |
| The loss of silver | 101 |
| Condition affecting the loss | 102 |
| Methods of correction | 103 |
| Lead required for cupellation | 105 |
| (4) Calculation of the results in ounces to the ton of 2240 lbs. Table | 107 |
| Ores with metallic particles | 108 |
| (5) Explanatory notes | 110 |
| (6) Examples of dry silver assays | 113 |
| Wet assays | 116 |
| Gravimetric method | 117 |
| Gay-Lussac's method | 119 |
| Volhard's method | 121 |
| A modified Gay-Lussac | 123 |
| Volhard's method applied to arsenic | 124 |
| Gold—Detection | 126 |
| Amalgamation assay | 126 |
| Dry assay | 127 |
| (1) Size of charges | 127 |
| (2) Sampling | 127 |
| (3) Assay tons | 131 |
| (4) Small buttons, weighing | 131 |
| " " measuring | 133 |
| (5) Concentration in lead | 136 |
| Quartz ores | 136 |
| Ores with oxide of iron | 138 |
| Ores with metallic sulphides | 139 |
| (6) Cyanide charges, residues, &c. | 140 |
| (7) Cupellation | 142 |
| Cupels | 142 |
| Cupellation temperature | 143 |
| Cupellation loss | 145 |
| (8) Inquartation | 146 |
| (9) Flatting | 149 |
| (10) Parting, in flasks | 151 |
| " in test tubes | 152 |
| " in glazed crucibles | 153 |
| " Loss, &c. | 154 |
| (11) Check assays, surcharge | 154 |
| (12) Bullion assays in special apparatus | 156 |
| Silver, &c., in gold bullion | 157 |
| (13) Sampling of base bullion, &c. | 157 |
| Cyanides—Commercial cyanides | 160 |
| Double cyanides | 161 |
| Prussic acid | 162 |
| Gold-dissolving power of cyanide liquor | 162 |
| Assay for cyanide strength | 163, 165 |
| Assay of commercial cyanide | 167 |
| Alkalinity of cyanides | 167 |
| Acidity of ores | 168 |
| Metals in cyanide liquors | 169 |
| Cyanicides | 169 |
| Platinum | 170 |
| Iridium | 171 |
| Mercury | 171 |
| Dry assay | 172 |
| Wet method | 173 |
| CHAPTER X. | |
| COPPER, LEAD, THALLIUM, BISMUTH, ANTIMONY. | |
| Copper—Introductory | 175 |
| Dry assay | 176 |
| Valuation of copper ores | 181 |
| Wet methods | 183 |
| (1) Electrolytic assay | 184 |
| Volumetric methods | 194 |
| (1) Cyanide method | 194 |
| (2) Iodide method | 199 |
| (3) Colorimetric method | 203 |
| Examination of commercial copper | 205 |
| Lead | 211 |
| Dry assay | 211 |
| Wet assay | 213 |
| (1) Gravimetric method | 213 |
| (2) Volumetric method | 214 |
| (3) Colorimetric method | 218 |
| Thallium | 219 |
| Bismuth | 220 |
| Dry assay | 221 |
| Wet method | 221 |
| (1) Gravimetric determination | 222 |
| (2) Colorimetric assay | 223 |
| Antimony | 225 |
| Dry assay | 225 |
| Wet method | 227 |
| (1) Gravimetric assay | 228 |
| (2) Volumetric method | 229 |
| CHAPTER XI. | |
| IRON, NICKEL, COBALT, ZINC, CADMIUM. | |
| Iron | 231 |
| Gravimetric determination | 233 |
| Permanganate and bichromate methods | 234 |
| Stannous chloride method | 244 |
| Colorimetric determination | 247 |
| Nickel | 251 |
| Dry assay | 251 |
| Electrolytic assay | 254 |
| Titration by cyanide | 255 |
| Cobalt | 259 |
| Zinc | 261 |
| Gravimetric method | 262 |
| Volumetric method | 263 |
| Gasometric method | 266 |
| Cadmium | 269 |
| CHAPTER XII. | |
| TIN, TUNGSTEN, TITANIUM. | |
| Tin | 271 |
| Vanning | 273 |
| Dry assay | 276 |
| Detection, &c. | 279 |
| Gravimetric determination | 281 |
| Volumetric determination | 282 |
| Examples | 284 |
| Titanium | 292 |
| Tungsten | 295 |
| Niobic and Tantalic Oxides | 297 |
| CHAPTER XIII. | |
| MANGANESE, CHROMIUM, ETC. | |
| Manganese | 298 |
| Gravimetric determination | 300 |
| Volumetric determination | 300 |
| Ferrous sulphate assay | 301 |
| Iodine assay | 302 |
| Colorimetric determination | 306 |
| Chromium | 307 |
| Vanadium | 310 |
| Molybdenum | 311 |
| Uranium | 312 |
| CHAPTER XIV. | |
| EARTHS, ALKALINE EARTHS, ALKALIES. | |
| Alumina | 314 |
| Thoria | 317 |
| Zirconia | 317 |
| Cerium | 318 |
| Lanthanum and Didymium | 319 |
| Yttria | 319 |
| Beryllia | 319 |
| Lime | 320 |
| Strontia | 324 |
| Baryta | 326 |
| Magnesia | 328 |
| The Alkalies | 330 |
| Sodium | 334 |
| Potassium | 336 |
| Lithium | 338 |
| Cæsium | 339 |
| Rubidium | 340 |
| Ammonium | 340 |
| PART III. | |
| CHAPTER XV. | |
| OXYGEN AND OXIDES—THE HALOGENS. | |
| Oxygen | 344 |
| Oxides | 345 |
| Water | 350 |
| The Halogens | 358 |
| Chlorine | 359 |
| Bromine | 361 |
| Iodine | 362 |
| Fluorine | 363 |
| CHAPTER XVI. | |
| SULPHUR AND SULPHATES. | |
| Sulphur | 367 |
| Gravimetric determination | 369 |
| Volumetric determination | 370 |
| Sulphates | 377 |
| Selenium | 379 |
| Tellurium | 379 |
| CHAPTER XVII. | |
| ARSENIC, PHOSPHORUS, NITROGEN. | |
| Arsenic | 381 |
| Gravimetric determination | 383 |
| Volumetric method, "iodine" | 384 |
| " " "uranic acetate" | 389 |
| Phosphorus | 394 |
| Gravimetric determination | 396 |
| Volumetric determination | 397 |
| Nitrogen and Nitrates | 400 |
| CHAPTER XVIII. | |
| SILICON, CARBON, BORON. | |
| Silicon and Silicates | 405 |
| Carbon and Carbonates | 414 |
| Coals | 418 |
| Shales | 420 |
| Carbonates | 424 |
| Boron and Borates | 429 |
| APPENDIX A. | |
| Table of atomic weights and other constants | 433 |
| Table for converting degrees of the centigrade thermometer | |
| into degrees of Fahrenheit's scale | 435 |
| Tables showing strengths of aqueous solutions of nitric and hydrochloric acids, | |
| of ammonia and of sulphuric acid | 436 |
| APPENDIX B. | |
| Estimation of small quantities of gold | 440 |
| Practical notes on the iodide process of copper assaying | 441 |
| Method of separating cobalt and nickel | 442 |
| APPENDIX C. | |
| A lecture on the theory of sampling | 444 |
| Index | 450 |
Assaying has for its object the determination of the quantities of those constituents of a material which add to or detract from its value in the arts and manufactures. The methods of assaying are mainly those of analytical chemistry, and are limited by various practical considerations to the determination of the constituents of a small parcel, which is frequently only a few grains, and rarely more than a few ounces, in weight. From these determinations calculations are made, which have reference to a mass of material of, perhaps, hundreds of tons. But in all cases, whether the mass under consideration be large or small, whether the material be obtained by mining, grown, or manufactured, the assayer is supposed to receive a small quantity, called "the sample," which is, or ought to be, the exact counterpart of the mass of material that is being dealt with. The taking and making of this sample is termed "sampling"; and the men whose special work it is to select such samples are "the samplers."
But although "sampling" is thus distinct from "assaying," the assayer should be familiar with the principles of sampling, and rigorous in the application of these principles in the selecting, from the sample sent him, that smaller portion upon which he performs his operations.
Sampling.—In the case of gases, there is absolutely no trouble in mixing. The only difficulty is in drawing off a fair sample where, as in flues, the body of the gas is in motion, and varies a little in composition from time to time. In this case, care must be taken to draw off uniformly a sufficient volume of the gas during a prolonged period; any portion of this larger volume may then be taken for the analytical operation.[Pg 2]
In the case of liquids, which mix more or less easily—and this class includes metals, &c., in the state of fusion—more or less severe agitation, followed by the immediate withdrawal of a portion, will yield a fairly representative sample.
In the case of solids, the whole mass must be crushed, and, if not already of fairly uniform quality, mixed, before sampling can take place. Most of the material which a sampler is called upon to deal with, is, however, in a more or less divided state and fairly uniform. In practice it is assumed that 5 per cent. of the whole (= 1/20th), if taken in portions of equal weight and at frequent and regular intervals, will represent the mass from which it was taken. Taking a heap of ore, A, and selecting one out of every twenty spade-, bag-, barrow-, or wagon-fuls, according to the quantity of stuff in the heap, there is obtained a second heap, B, containing one-twentieth of the stuff of the heap A. If we crush the stuff in B until this heap contains approximately the same number of stones as A did—which means, crushing every stone in B into about twenty pieces—B will become the counterpart of A. Selecting in the same manner 5 per cent. of B, there is got a third heap, C. This alternate reduction and pulverising must be carried on until a sample of suitable size is obtained. This may be expressed very clearly thus:—
A = 1000 tons of rocks and lumpy ore.
B = 50 " " rough stones, 1/20th of A.
C = 2.5 " " small stones, 1/20th of B.
D = 0.125 " " coarse powder, 1/20th of C.
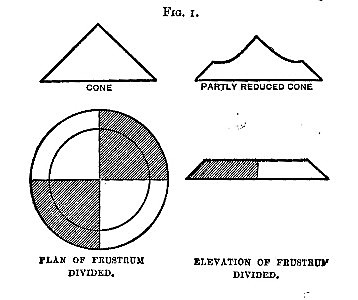
If the material to be sampled is already a dry powder, 5 per cent. of it should be heaped in a cone; each lot being added on[Pg 3] the apex of the cone already formed, so that it may distribute itself by falling evenly in all directions. When the cone is completed, convert it into a low frustrum of a cone by drawing stuff uniformly and in a direct line from the centre to the circumference. Draw two diameters at right angles to each other, and reserving any two alternate quarters, reject the others. Mix; and form another cone, and proceed until a sample is got of the bulk required.
This is the usual plan, and all samples should be treated in this way when the stuff is fine enough to fall evenly down the sides of a cone.
Samples as they reach the assay office are seldom in a fit state for the work of the assayer; they are generally too coarse, and ought always to be more than he wants for any particular determination. The portion he requires should never be taken at hap-hazard; the sample must be reduced systematically to the quantity required.
1. If the sample is a liquid: it is sufficient to shake the bottle, and take out a measured or weighed quantity for the assay.
2. If a liquid with a solid in suspension: measure the whole of it. Filter. Make up the filtrate with the wash-water or water to the original bulk. Assay it. Dry and weigh the residue, and make a separate assay of it.
3. If of a creamy consistency, free from heavy particles: mix well; spread out evenly on a glazed tile. Take up equal portions at equal distances. Mix and assay.
4. If a mud of coarse and fine particles, or of particles of unequal density: weigh and transfer to a porcelain dish, or weigh in the dish. Dry at 100° C., weigh. Treat the residue as a solid capable of being powdered.
5. If a solid capable of being powdered, or already powdered: heap up into a cone; flatten with a spatula; divide along two diameters at right angles, and carefully reject the whole of two alternate quarters, brushing away any fine powder. Mix the other quarters, and repeat (if necessary). For small quantities a fine state of division is essential.
6. If a solid with metallic particles: powder and pass through a sieve; the metallic particles will not pass through. Weigh both portions and assay separately. Sifting should be followed by a very thorough mixing.
7. If a metal or alloy in bar or ingot: clean the upper surface of the bar, and bore through the bar. Use the borings. If the ingot or bar is small, cut it through and file the section. Filings must be freed from fragments of the file by means of a magnet; and from oil, if any be present, by washing with a suitable[Pg 4] solvent.[1] Where practicable, metals and alloys are best sampled by melting and granulating. The student must carefully avoid any chance of mixing dirt or particles of other samples with the particular sample which he is preparing. One ore should be done at a time, and when finished, it should be labelled and wrapped up, or bottled, before starting on a fresh sample.
When an ore requires to be very finely ground in an agate mortar, it is often advisable to mix with a little pure alcohol and rub until free from grit; dry at 100° C. and mix well before weighing.
When an assay is required of a quantity of ore made up of parcels of different weight and quality, each parcel should be separately sampled and parts of each sample, bearing to each other the same proportion by weight as the original parcels, should be taken and mixed. For example, a lot of ore is made up of one parcel of A, 570 tons, one of B, 180 tons, and another of C, 50 tons; a sample representing the whole may be got by mixing 57 parts of a sample of A with 18 parts of a sample of B, and 5 parts of a sample of C.
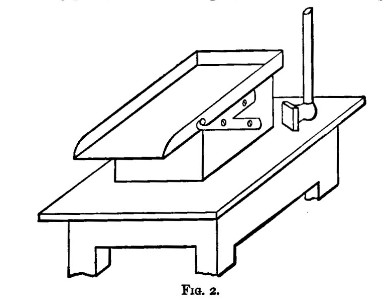
A bruising plate, like that in fig. 2, is convenient for general office work. The slab is of cast iron, about an inch thick. It is firmly supported on a solid block of wood, and pivoted for convenience in emptying. The bruising-hammer is steel-faced, about 4 inches square, and 1-1/2 inch thick. The block is firmly fixed to a small table or tressel, so that the slab is about 2 feet 6 inches[Pg 5] from the ground. The slab is cleaned, and the sample collected with the help of a stiff-haired brush.
Drying: Determination of Moisture.—In practice, the moisture is generally determined by the samplers, and the proportion is specified in grains per pound on the label attached to the sample when it reaches the assay office. The method adopted is usually to dry 1 lb. = 7000 grs. of the ore in a frying-pan heated over a gas flame, or in an ordinary oven, until a cold bright piece of metal or glass is no longer damped when held over it. The loss of weight in grains = moisture.
Properly, however, this work should be done by the assayer, if only for the following reason. It is assumed that the dry ore of the sampler and of the assayer are the same thing; according to the nature of the ore, this may or may not be the case. The assayer, however, uses the sample which he has dried for his moisture-determination, as the dry ore on which he makes his other assays, and no variation in moisture would influence the other and more important determinations. Some ores are sent to the smelter with from 5 to 15 per cent. of adherent water. In these cases it is best to spread out the sample, and taking equal portions fairly at regular intervals, weigh into a Berlin dish 20 grams. This should then be dried over a sand-bath, or if the ore is likely to be injured by excess of heat, over a water-bath until the weight is constant. The loss of weight multiplied by 5 gives the percentage of water present.
Example:—
| Weight of dish + wolfram | 32.67 | grms. |
| " " dish | 12.67 | " |
| ——— | ||
| " " wolfram | 20.00 | " |
| " " dish + wolfram | 32.67 | " |
| " " " dried | 30.15 | " |
| ——— | ||
| " " water | 2.52 | " |
| 2.52 × 5 = 12.6 | 12.6%. |
There are other ores which are not apparently wet, but in the state called "air-dried." It is easier to take fair samples of these, and, consequently, it is not necessary to use so large a quantity as 20 grams. But with a smaller quantity, extra precautions must be taken. All dry solids at ordinary temperatures absorb moisture from the air. The amount varies with the nature of the material and with the quantity of surface exposed. Light bulky powders absorb more than heavy ones, because of the greater condensing surface. It is on this account that it is well to weigh substances, which have been dried, between close-fitting watch-glasses. The[Pg 6] method of determining moisture is to weigh out into the glasses 5 grams of ore, and dry in the water-oven until there is no further loss of weight. On taking the glasses out of the oven, they should be at once closed, the clip put on, and after cooling in a desiccator weighed. If after a second trial the loss is the same, or only increased by a milligram, the determination is finished.
Example:—
| Weight of glasses + pyrites | 31.0470 | grms. |
| " " glasses | 26.0470 | " |
| ——— | ||
| " " pyrites | 5.0000 | " |
| " " glasses + pyrites, dried 1 hour | 30.8965 | " |
| " " " " dried 1-1/2 " | 30.8957 | " |
| " " " " | 31.0470 | " |
| " " " " dried | 30.8957 | " |
| ——— | ||
| " " moisture | 0.1513 | " |
| 0.1513 × 20 = 3.026 | 3.02%. |

Sometimes it may be advisable to dry 10 grams, in which case multiplying the loss by 10 will give the percentage. The dried ore should be transferred to a weighing-tube (fig. 3), and reserved for the subsequent determinations. The weighing-tube with the ore must be marked, and kept in a desiccator.
Most ores and inorganic substances can be dried, and their moisture determined by the loss in this way. When, however, the substance contains another somewhat volatile ingredient, it is exposed over sulphuric acid in a desiccator for two days (if in vacuo, all the better), and the loss determined. Moisture in dynamite should be determined in this way.
When water is simply mechanically mixed with a substance it presents but little difficulty. The combined water is a different matter. Slaked lime, even when perfectly dry, contains much water; and if the water of soda crystals were separated and frozen, it would occupy a volume equal to that of the original crystals. Perfectly dry substances may contain much water, and this combined water is retained by different materials with very unequal vigour. Sodium sulphate and sodium phosphate crystals lose water even when exposed under ordinary conditions to dry air. Soda crystals when heated melt, and at a moderate temperature give off their water with ebullition. The temperature at which all the water is given up varies with each particular salt; the actual determination of the water in each case will require somewhat different treatment. Such determinations, however, are seldom[Pg 7] required; and from a practical point of view this combined water causes no trouble.
In assaying ores, we term "moisture" all water which is lost by exposure in a water-oven at 100° C., and the "dry ore" is the ore which has been dried at this temperature. No advantage, but rather endless confusion, would be caused by varying the temperature with the object of estimating the whole of the water which a hydrated salt may contain. The results of the assay of the other components should be calculated on the "dry ore." One advantage of this is obvious:—The dry ore has a constant composition, and the results of all assays of it will be the same, no matter when made; the moisture, however, may vary from day to day, and would be influenced by a passing shower of rain. It is well to limit this variability to the moisture by considering it apart, and thus avoid having the percentage, say, of copper rising and falling under the influence of the weather.
In the case of certain salts, however, such as soda crystals and hydrated sulphate of copper (when these constitute the bulk of the substance to be assayed), it is as well to perform the assay on the moist, or at any rate air-dried, substance.[2] It would be equally convenient to calculate on the substance dried at 100° C.; but in this case it would be well, in order to avoid a somewhat shallow criticism, to replace the term "moisture" by the longer but equivalent phrase "water lost at 100° C."
Calculation and Statement of Results.—By far the most generally convenient method of stating the results of an assay is that of the percentage or parts in a hundred, and to avoid a needlessly troublesome calculation it is well to take such a quantity of ore for each assay as by a simple multiplication will yield the percentage. In these calculations decimals are freely employed, and students should make themselves familiar with the methods of using them.
Other methods of statement are in use, and have advantages in certain special cases. With bullion the parts in a thousand are given, and in those cases in which the percentage is very small, as in water analysis, it is convenient to report on parts in 100,000, or even on parts per 1,000,000. These are easily got from the corresponding percentages by shifting the decimal point one, three, or four places to the right. Thus 92.5 per cent. is 925 per thousand; and 0.0036 per cent. is 3.6 per 100,000, or 36 per million.
With ores of tin, silver, and gold, the result is stated as so many cwts., lbs., or ozs., in the ton. With dressed tin ores as they are[Pg 8] sent to the smelter, the produce is given in cwts. and quarters to the ton. The corresponding percentage may be obtained by multiplying by five; or, inversely, if the percentage is given, the produce may be got by dividing by five. A produce of 13-1/2 equals a percentage of 13.5 × 5 = 67.5; and a percentage of 70.0 equals a produce of 70 / 5 = 14. With tin ores as raised (in which the percentage is small) the reduction must be carried to pounds per ton. One per cent. equals 22.4 lbs. to the ton; consequently, if we multiply the percentage by 22.4, the produce will be given. Thus, if an ore contains 6.7 per cent. of oxide of tin, the produce is 6.7 × 22.4 = 150 lbs. (or 1 cwt., 1 quarter, and 10 lbs.) to the ton. With gold and silver ores, the proportion of precious metal is small, and it is necessary to carry the reduction to ozs. and dwts. to the ton; and since gold and silver are sold by troy weight, whilst the ton is avoirdupois, it is of importance to remember that the ounces in the two systems are not the same. A ton contains 15,680,000 grains, which equal 653,333.3 dwts. or 32,666.6 ozs. (troy). The following rules are useful:—
To get ozs. (troy) per ton, multiply parts per 100,000 by 0.327;
To get dwts. per ton, multiply parts per 100,000 by 6.53;
To get grains per ton, multiply parts per 100,000 by 156.8.
Where liquids are being assayed, cubic centimetres are held to be equivalent to grams, and the usual method of statement is, "so many parts by weight in so many by measure." Where the statement is made as grams per litre or grains per gallon, there can be no doubt as to what is meant; and even if it be expressed in parts per 100,000, parts by weight in a measured volume must be understood unless the contrary is expressly stated.
In some cases, where the density of the solution differs greatly from that of water, the percentage by weight may be given; and in others, mixtures of two or more liquids, the percentages may be given by volume or by weight; as so many c.c. in 100 c.c., or as so many grams in 100 grams, or even as so many grams in 100 c.c. In such cases it must be distinctly shown which method of statement is adopted.
One grain per gallon means 1 grain in 70,000 grain-measures, or one part in 70,000. Dividing by 7 and multiplying by 10 will convert grains per gallon into parts per 100,000. Inversely, dividing by 10 and multiplying by 7, will convert parts per 100,000 into grains per gallon.
Grams per litre are parts per 1000; multiplying by 100 will give parts per 100,000, and multiplying by 70 will give grains per gallon.
Among foreign systems of weights, the French is by far the best. Kilograms (2.205 lbs.) per quintal (220.5 lbs.) are parts per cent.; and grams (15.43 grs.) per quintal are parts per[Pg 9] 100,000. From the rule already given, grams per quintal may be converted into ounces to the ton by multiplying by 0.327.
The German loths per centner (1/2 oz. (avoirdupois) to 100 lbs.) equal parts per 3200; they are converted into parts per cent. by dividing by 32, or into ounces (troy) per ton by multiplying by 10.208.
In the United States, as a sort of compromise between the avoirdupois and metric systems, a ton is taken as 2000 lbs. There, too, the custom is adopted of reporting the gold and silver contents of an ore as so many dollars and cents to the ton. In the case of gold, an ounce is considered to be worth 20.6718 dollars. With silver, the nominal value is 1.2929 dollars per ounce, but frequently in assay reports it is taken as one dollar. The practice is objectionable. The prices of metals vary with the fluctuations of the market, and if the assayer fixed the price, the date of his report would be all important; if, on the other hand, he takes a fixed price which does not at all times agree with the market one, it leaves a path open for the deception of those unacquainted with the custom. American "dollars on the ton of 2000 lbs." may be converted into "ounces in the ton of 2240 lbs." by dividing by 1.1544 in the case of silver, and by 18.457 in the case of gold.
Laboratory Books and Report Forms.—The record which the assayer makes of his work must be clear and neat, so that reference, even after an interval of years, should be certain and easy. One method should be adopted and adhered to. Where there are a large number of samples, three books are required.
Sample Book.—This contains particulars of the samples (marks, &c.), which are entered by the office-clerk as they arrive. He at the same time puts on each sample the distinguishing number.
Example of Page of Sample Book.
| Date. | Number. | Sample. | Remarks. |
| Feb. 1 | 482 | Tough Copper | For Arsenic. |
| " 2 | X | Piece of Metal | For Ni and Cu. |
| " | 483 | Tough Copper. | |
| " | 73 | Silver Precipitate, 4 casks, 24 cwt. 1 qr. | With Letter. |
| " | 494 | Purple Ore, 200 tons. | |
| " | 1 J.T. | Lead Ore, 1 J.T. | From Corsica. |
| " | 2 J.T. | " 2 J.T. |
Laboratory Book. This is the Assayer's note-book, in which he enters clearly the particulars of his work—the results obtained, as[Pg 10] well as how these results were arrived at. The calculations should be done on scrap-paper, and should not be entered, although, of course, detail enough must be shown to enable the results to be recalculated.
Example of Page of Laboratory Book.
____________________________________________________________
Purple Ore 5 grams
19/10/89 0.0042 grm.
0.0021 "
———
Colorimetric 0.0063 × 20 = 0.13% Copper
______________________________________________________________
482
Tough Copper 10 grams
Feb. 1/89 10.5 c.c. Uranium.
= 0.52% Arsenic
______________________________________________________________
2082
Tough Copper 10 grams
12.7 c.c. Uranium.
= 0.63% Arsenic
______________________________________________________________
491 10 grams
Tough Copper 13.7 c.c. Uranium
Feb. 1/89
= 0.68% Arsenic
______________________________________________________________
Standard of Uranium acetate.
0.150 gram As2O3 = 23.3 c.c. Uranium.
∴ 100 cc. Uranium = 0.5 gram As.
______________________________________________________________
10071 5 grams
Tin Ore Cruc. and SnO2 9.6065 grms.
Feb. 3/89 Cruc. and Ash 9.4235 "
———
SnO2 = 0.1830 = 2.88% Tin
______________________________________________________________
The Assay Book.—This is the Official book, and is a combination of the Sample and Laboratory books. It corresponds with the report-forms. Without being loaded with detail, it should contain sufficient to characterise each sample.[Pg 11]
Example of Page of Assay Book.
Description of Sample.
| Date. | Material. | Weight. | No. | Water Lost at 100° C. | Assay on the Dry Material. | Date Reported. | ||||
| 1889 | ton | cwt | qrs | lbs | ||||||
| Feb. 1 | Tough cake copper | ... | ... | ... | ... | 482 | ... | Arsenic, 0.52% | 7 | |
| " | Tough cake copper | ... | ... | ... | ... | 2082 | ... | Arsenic, 0.63% | 7 | |
| " | Tough cake copper | ... | ... | ... | ... | 491 | ... | Arsenic, 0.68% | 7 | |
| Feb. 2 | Nickel disc for C.R. | ... | ... | ... | ... | X | ... | Copper, 73.75 | 7 | |
| Nickel, 24.34 | ||||||||||
| Iron, 2.18 | ||||||||||
| ——— | ||||||||||
| 100.27 | ||||||||||
| ——— | ||||||||||
| " | Silver precipitate, 4 casks | ... | 24 | 1 | 0 | 73 | Not det. | Silver, 4.851 | 10 | |
| Gold, 0.0215 | ... | |||||||||
| Lead, 19.37 | ... | |||||||||
| Zinc, 2.00 | ... | |||||||||
| Silver, 1584.7 ozs. per ton | ... | |||||||||
| Gold, 7.0 ozs. per ton | ... | |||||||||
| " | Purple ore | ... | 200 | ... | ... | ... | 494 | Not det. | Copper, 0.13% | 11 |
| Sulphur 0.15% | ... |
When the number of samples is small, the Sample Book may be omitted, and the entries made in the Assay Book as the samples arrive.
Report-forms. These should entail as little writing as possible in making out the report. For general purposes the form given on p. 12 is useful.
The quantity of substance to be taken for any particular assay depends largely upon the method of assay adopted. There are, however, some general considerations which should be remembered, and some devices for simplifying the calculations which should be discussed.
The smaller the percentage of the substance to be determined, the larger should be the amount of the ore taken. The following table will give a general idea as to this:—
| Percentage of the substance to be determined. | Amount of ore, &c. to be weighed. |
| 100-10 | 1 gram. |
| 10-5 | 2 grams. |
| 5-1 | 5 " |
| 1-0.1 | 10 " |
| 0.1-0.01 | 20 " |
 ASSAY NOTE
ASSAY NOTE
The rougher the method of assay adopted, the larger should be the[Pg 13] quantity of ore taken. If the degree of accuracy attainable with the methods and instruments at the assayer's service is known, it is easy to calculate what quantity should be taken for any particular case. If the results are good within 0.001 gram, then, taking 1 gram of ore we can report within 0.1 per cent., or if they are good within 0.0002 gram, taking 20 grams of ore, we can report within 1 part per 100,000, or very closely within 6-1/2 dwt. to the ton. If it is wished to be yet more particular in reporting, larger quantities must be taken. The difficulty of manipulating very small or very large precipitates, &c., must be borne in mind. So, too, must the fact that the greater the weight of the final product of an assay, the less, as a rule, is the percentage error. The distinction between absolute and percentage error, often overlooked, is important. If 0.5 gram of silver be cupelled with 20 grams of lead, there may be obtained a button of 0.495 gram; the absolute loss is 0.005 gram, and this equals 1 per cent. of the silver present. Similarly, cupelling 0.1 gram, the resulting button may be 0.098; the absolute loss is only 0.002 gram, but this equals 2 per cent. of the silver present. In the same way the student should see that the two results, 91.5 per cent. and 92.0 per cent., are really more concordant than the results 9.1 per cent. and 9.2 per cent.
A device often adopted in practice where a large number of assays of one kind are made, and the report is given as so many ounces or pounds to the ton, is that known as the assay ton. The assay ton may be any arbitrary and convenient weight, but its subdivisions must bear to it the same relations as pounds and ounces bear to the actual ton. On the other hand, in a laboratory where many kinds of work are performed, different sets of weights of this kind would only tend to confusion, even if they were not unnecessary. With a set of gram weights and its subdivisions anything may be done. If it is desired to report as pounds to the ton, then, since there are 2240 lbs. to the ton, a weight of 2.240 grams may be taken as the assay ton, and each 0.001 gram yielded will equal 1 lb., or 22.4 grams may represent the ton, and each 0.01 gram a pound. Similarly, since there are 32,666.6 ozs. troy to the ton; if we take 32.6667 grams as the assay ton, each 0.001 gram will equal 1 oz. to the ton. In some cases it may be convenient to have, in addition to the usual gram weights, one or other of the "assay tons" mentioned above, but generally it is better to work on a purely decimal system, and convert when required into ounces per ton, &c., either by actual calculation or by reference to a set of tables.[Pg 14]
Practical Exercises.
The student should practise such calculations as the following:—
1. Calculate the percentages in the following cases:—
(a) Ore taken, 2 grams; copper found, 0.2155.
(b) " 1.5 gram; iron found, 0.8340.
(c) " 30 grams; lead found, 23.2.
2. Calculate the parts per thousand in the following:—
(a) Bullion taken, 1.1 gram; silver found, 1.017.
(b) " 1.14 gram; silver found, 1.026.
(c) " 0.6 gram; gold found, 0.5500.
3. Calculate parts per 100,000 in the following:—
(a) Ore taken, 20 grams; silver found, 0.0075.
(b) " 50 grams; gold found, 0.0026.
(c) Water taken, 500 c.c.; solids found, 0.1205.
4. Calculate cwts. to the ton in the following:—
(a) Ore taken, 5 grams; tin found, 2.816.
(b) " 5 grams; tin found, 3.128.
(c) An ore with 68.2 per cent. of tin.
5. Calculate lbs. to the ton in the following:—
(a) An ore with 3.28 per cent. oxide of tin.
(b) Ore taken, 20 grams; oxide of tin found, 1.67.
6. Calculate ozs. (troy) to the ton in the following:—
(a) Ore taken, 50 grams; gold found, 0.0035.
(b) " 20 grams; silver found, 0.0287.
(c) " 25 grains; silver found, 0.0164.
7. Calculate in grains per gallon:—
(a) 0.51 gram per litre.
(b) 24.6 parts per 100,000.
(c) Solution taken, 100 c.c.; copper found, 0.0045 gram.
(d) " 50 c.c.; iron found, 0.165 gram.
8. Convert into ozs. (troy) per ton:—
(a) 7 loths per centner.
(b) 30 grams per quintal.
(c) 15 parts per 100,000.
[1] Ether or carbon bisulphide.
[2] Such substances are best dried by pressing between folds of dry filter-paper.
The methods of assaying are best classed under two heads, Gravimetric and Volumetric, in the former of which the final results are weighed, whilst in the latter they are measured. A commoner and older division is expressed in the terms much used in practice—wet assays and dry assays. Wet assays include all those in which solvents, &c. (liquid at the ordinary temperature), are mainly used; and dry assays, those in which solid re-agents are almost exclusively employed. Dry assays form a branch of gravimetric work, and we shall include under this head all those assays requiring the help of a wind furnace. Wet assays, as generally understood, would include not only those which we class as wet gravimetric assays, but also all the volumetric processes.
Gravimetric Methods aim at the separation of the substance from the other matters present in the ore, so that it may be weighed; and, therefore, they must yield the whole of the substance in a pure state. It is not necessary that a metal should be weighed as metal; it may be weighed in the form of a compound of definite and well known composition. For example, one part by weight of silver chloride contains (and, if pure, always contains) 0.7527 part of silver; and a quantity of this metal can be as exactly determined by weighing it as chloride as by weighing it in the metallic state. But in either case the metal or its chloride must be pure.
Exact purity and complete separation are not easily obtained; and methods are used which are defective in one or both of these respects. It is well to note that an impure product increases the result, whilst a loss of the substance decreases it; so that if both defects exist in a process they tend to neutralise each other. Of dry methods generally, it may be said that they neither give the whole of the substance nor give it pure; so that they are only calculated to show the amount of metal that can be extracted on a manufacturing scale, and not the actual quantity of it present.[Pg 16] Their determinations are generally rough and always low. The gold and silver determinations, however, will compare very favourably with any of the other processes for the estimation of these metals in their ores.
The calculation of the results of a gravimetric assay has already been referred to. If the result is to be stated as percentage, it may always be done by the following rule:—Multiply the weight of the substance got by the percentage of metal it contains, and divide by the weight of ore taken.
Gravimetric methods are divided into three groups: (1) mechanical separations; (2) dry methods; and (3) wet methods.
Mechanical Separations.—Under this head are classed the method of assaying tin ores, known as vanning, and the amalgamation assay for gold. A set of sieves to determine the relative proportion of powders of different degrees of fineness is sometimes useful. A set with 10, 20, 40 and 80 meshes to the inch is convenient.
Dry Assays.—An important distinction between wet and dry methods of assaying is, that in the former the substance is got into the liquid state by solution, whilst in the latter fusion is taken advantage of.
The difference between solution and fusion is easily illustrated: a lump of sugar heated over a candle-flame melts or fuses; suspended in water it dissolves. Many substances which are insoluble or infusible of themselves, become soluble or fusible when mixed with certain others; thus, in this way, solution is got with the aid of reagents, and fusion with the help of fluxes. For example, lead is insoluble in water, but if nitric acid be added, the metal rapidly disappears. It is convenient, but somewhat inaccurate, to say that the acid dissolves the lead. If the lead be acted on by nitric acid alone, without water, it is converted into a white powder, which does not dissolve until water is added; in this case it is obvious that the water is the solvent. The function of the acid is to convert the lead into a soluble compound.
Fluxes may act as true solvents. Fused carbonate of soda dissolves baric carbonate, and perhaps in many slags true solution occurs; but in the great majority of cases a flux is a solid reagent added for the purpose of forming a fusible compound with the earthy or stony minerals of the ore. Few of the minerals which occur in the gangue of an ore are fusible; and still fewer are sufficiently fusible for the purposes of the assayer, consequently the subject is one of importance, and it ought to be treated on chemical principles. An idea of the composition of some of the more frequently occurring rocks may be gathered from the following table, which represents rough averages:[Pg 17]—
| Silica. | Alumina. | Oxide of iron | Lime and Magnesia. | Alkalies. | |
| % | % | % | % | % | |
| Sandstone, grit, quartzite, &c. | 80-100 | — | — | — | — |
| Granite, gneiss, quartz-porphyry, fire-clay, &c. | 70-75 | 13-20 | 2 | 2 | 5-8 Less in fire-clay. |
| Mica-schist | 65 | 18 | 5 | 3 | 3 |
| Trachyte, syenite | 60 | 17 | 7 | 4-7 | 6-9 |
| Clay-slate | 60 | 18 | 10 | 8 | 3 |
| Diorite | 54 | 17 | 12 | 9 | 3-4 |
| Horneblende-rock | 50 | 18 | 15 | 12 | 3-4 |
| Brick-clay | 50 | 34 | 8 | 6 | — |
| China-clay | 47 | 39 | — | — | — |
| Basalt, dolerite, &c. | 50 | 15 | 15 | 16 | 3 |
| Serpentine | 44 | — | — | 44 | — |
| Chalk, limestone, dolomite, &c. | — | — | — | 45-55 | — |
Silica itself, and the silicates of alumina, of lime, and of magnesia, are practically infusible; the silicates of soda, of potash, and of iron are easily fusible if the base (soda, potash, or oxide of iron) be present in sufficient quantity, and if, in the case of the iron, it is present mainly as lower oxide (ferrous silicate). The addition of lime, oxide of iron, or alkali to silicate of alumina results in the formation of a double silicate of alumina and lime, or of alumina and iron, &c., all of which are easily fusible. Similarly, if to a silicate of lime we add oxide of iron, or soda, or even alumina, a fusible double silicate will be formed. Thus lime, soda, oxide of iron, and clay, are fluxes when properly used; but since lime, clay (and oxide of iron if there be any tendency to form peroxide), are of themselves infusible, any excess of these fluxes would tend to stiffen and render pasty the resulting slag. So, too, soda, which is a very strong base, may act prejudicially if it be in sufficient excess to set free notable quantities of lime and magnesia, which but for that excess would exist in combination as complex fusible silicates. There are many minerals which with but little soda form a glass, but with more yield a lumpy scoriacious mass. There are many minerals, too, which are already basic (for example, calcite), and which, when present, demand either a less basic or an acid flux according to the proportions in which they exist. For purposes of this kind borax, or glass, or clay with more or less soda may be used, and of these borax is by far the most generally useful. An objection to too basic a slag (and a very important one) is the speed with which it corrodes[Pg 18] ordinary crucibles. These crucibles, consisting of quartz and clay, are rapidly attacked by lime, soda and bases generally.
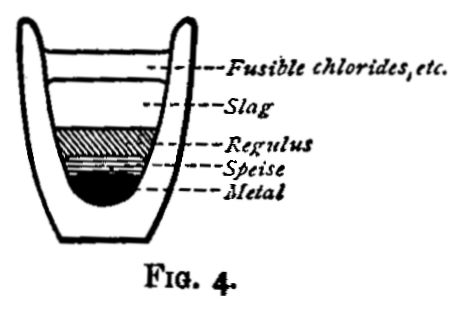
In considering what is and what is not a good slag, certain chemical properties are of importance. If a mixture of many substances be fused and allowed to solidify in a crucible, there will be found some or all of the following. At the bottom of the crucible (fig. 4) a button of metal, resting on this a speise; then a regulus, next a slag made up of silicates and borates and metallic oxides, and lastly, on the top another layer of slag, mainly made up of fusible chlorides and sulphates. In assaying operations the object is generally to concentrate the metal sought for in a button of metal, speise or regulus, and to leave the earthy and other impurities as far as possible in the slag; whether there be one or two layers of slag is a matter of indifference;[3] but the chemical action of the lower layer upon the speise, or regulus, or metal, is of great importance.
A regulus is a compound of one or more of the metals with sulphur; it is usually brittle, often crystalline, and of a dull somewhat greasy lustre. It is essential that the slag, when solid, shall be so much more brittle than the regulus, that it shall be easy to crumble, and remove it without breaking the latter; and it must not be basic. The effect of fusing a regulus with a basic slag is well seen when sulphide of lead is fused with carbonate of soda; the result is a button of metal (more or less pure), and a slag containing sulphides of lead and sodium; and again, if sulphide of lead be fused with an excess of oxide of lead, a button of lead will be got, and a slag which is simply oxide of lead (with whatever it may have taken up from the crucible), or if a sufficient excess has not been used, oxide of lead mixed with some sulphide. When (as is most frequently the case) the desire is to prevent the formation of regulus, these reactions may be taken advantage of, but otherwise the use of a flux having any such tendency must be avoided. A good slag (from which a regulus may be easily separated) may be obtained by fusing, say, 20 grams of ore with borax 15 grams, powdered glass 15 grams, fluor spar, 20 grams, and lime 20 grams; by quenching the slag in water as soon as it has solidified, it is rendered very brittle.
Sulphide of iron formed during an assay will remain diffused[Pg 19] through the slag, instead of fusing into a button of regulus, if the slag contain sulphide of sodium. The same is true of other sulphides if not present in too great a quantity, and if the temperature is not too high.
Speises are compounds of a metal or metals with arsenic. They are chiefly of interest in the metallurgy of nickel, cobalt, and tin. They are formed by heating the metal or ore in covered crucibles with arsenic and, if necessary, a reducing agent. The product is fused with more arsenic under a slag, consisting mainly of borax. They are very fusible, brittle compounds. On exposure to the air at a red heat the arsenic and the metal simultaneously oxidize. When iron, cobalt, nickel, and copper are present in the same speise, they are eliminated in the order mentioned.
Slags from which metals are to be separated should not be too acid; at least, in those cases in which the metal is to be reduced from a compound, as well as separated from earthy impurities. Where the object is simply to get a button of metal from a substance in which it is already in the metallic state, but mixed with dross (made up of metallic oxides, such as those of zinc or iron), from which it is desired to separate it, an acid flux like borax is best; or, if the metal is easily fusible, and there would be danger of loss of metal by oxidation or volatilising, it may be melted under a layer of resin or fat. Common salt is sometimes used with a similar object, and is often useful. Under certain conditions, however, it has a tendency to cause the formation of volatile chlorides with a consequent loss of metal.
In the great majority of cases, the fusion of the metal is accompanied by reduction from the state of oxide; in these the slag should be basic. It is not easy to reduce the whole of a reducible oxide (say oxide of copper or of iron) from a slag in which it exists as a borate or silicate; there should be at least enough soda present to liberate it. When the object is to separate one metal, say copper, without reducing an unnecessary amount of another (iron) at the same time, a slag with a good deal of borax is a distinct advantage. The slag then will probably not be free from copper, so that it will be necessary to powder and mix the slag with some soda and a reducing agent, and to again fuse the slag in order to separate this residual metal. In all those cases in which the slag retains an oxide of a heavy metal, this cleaning of the slag is advisable, and in the case of rich ores necessary. Slags containing sulphides are especially apt to retain the more easily reducible metals.
The following are the ordinary and most useful fluxes:—
Soda.—The powdered bicarbonate, sold by druggists as "carbonate[Pg 20] of soda," is generally used. It gives off its water and excess of carbonic acid readily and without fusion. Where the melting down is performed rapidly, the escaping gas is apt to cause trouble by frothing, and so causing waste of the material. Ordinary carbonate of soda, when hydrated (soda crystals), melts easily, and gives off its water with ebullition. It is unfit for use in assaying, but when dried it can be used instead of the bicarbonate. One part of the dried carbonate is equivalent to rather more than one and a half parts of the bicarbonate. From two to four parts of the flux are amply sufficient to yield a fluid slag with one part of earthy matter. This statement is also true of the fluxes which follow.
Borax is a hydrated biborate of soda, containing nearly half its weight of water. When heated it swells up, loses its water, and fuses into a glass. The swelling up may become a source of loss in the assay by pushing some of the contents out of the crucible. To avoid this, fused or dried borax may be used, in which case a little more than half the amount of borax indicated will suffice. Borax will flux almost anything, but it is especially valuable in fluxing lime, &c., and metallic oxides; as also in those cases in which it is desired to keep certain of the latter in the slag and out of the button of metal.
Oxide of Lead, in the form of red lead or litharge, is a valuable flux; it easily dissolves those metallic oxides which are either infusible or difficultly fusible of themselves, such as oxides of iron or copper. The resulting slag is strongly basic and very corrosive; no crucible will long withstand the attack of a fused mixture of oxides of lead and copper. With silicates, also, it forms very fusible double silicates; but in the absence of silicates and borates it has no action upon lime or magnesia. Whether the lead be added as litharge or as red lead, it will exist in the slag as monoxide (litharge); the excess of oxygen of the red lead is thus available for oxidising purposes. If this oxidising power is prejudicial, it may be neutralised by mixing the red lead with 1 per cent. of charcoal.
Glass: broken beakers and flasks, cleaned, dried, and powdered will do. It should be free from lead.
Fluor: fluor-spar as free as possible from other minerals, powdered. It helps to flux phosphate of lime, &c., and infusible silicates.
Lime: should be fresh and powdered. It must not be slaked. Powdered white marble (carbonate of lime) will do; but nearly double the quantity must be taken. One part of lime produces the same effect as 1.8 parts of the carbonate of lime.
Tartar and "black flux," are reducing agents as well as fluxes.[Pg 21] The "black flux," which may be obtained by heating tartar, is a mixture of carbonate of potash and charcoal.
Reducing Agents.—The distinction between reducing agents and fluxes (too often ignored) is an important one. Fluxes yield slags; reducing agents give buttons of regulus or of metal. The action of a reducing agent is the separation of the oxygen or sulphur from the metal with which it is combined. For example, the mineral anglesite (lead sulphate) is a compound of lead, sulphur, and oxygen; by carefully heating it with charcoal the oxygen is taken away by the charcoal, and a regulus of lead sulphide remains. If the regulus be then fused with metallic iron the sulphur is removed by the iron, and metallic lead is left. The charcoal and the iron are reducing agents. But in defining a reducing agent as one which removes oxygen, or sulphur, from a metallic compound so as to set the metal free, it must be remembered that sulphur itself will reduce metallic lead from fused litharge, and that oxygen will similarly set free the metal in fused lead sulphide. There is no impropriety in describing sulphur as a reducing agent; but it is absurd to call oxygen one. Some confusion will be avoided if these substances and those which are opposite to them in property be classed as oxidising and de-oxidising, sulphurising, and de-sulphurising agents. Most oxidising agents also act as de-sulphurisers.
The de-oxidising agents most in use are the following:—
Charcoal.—Powdered wood charcoal; it contains more or less hygroscopic moisture and about 3 or 4 per cent. of ash. The rest may be considered carbon. Carbon heated with metallic oxides takes the oxygen; at low temperatures it forms carbon dioxide, and at higher ones, carbon monoxide. Other conditions besides that of temperature have an influence in producing these results; and as the quantity of charcoal required to complete a definite reaction varies with these, it should be calculated from the results of immediate experience rather than from theoretical considerations.
Flour.—Ordinary wheat flour is convenient in use. On being heated it gives off inflammable gases which have a certain reducing effect, and a residue of finely divided carbon is left. It is likely to vary in the quantity of moisture it contains. Two parts of flour should be used where one part of charcoal would be otherwise required.
Tartar.—This is crude hydric potassic tartrate; the purified salt, cream of tartar, may be used. On being heated it gives off inflammable gases, and leaves a residue formed of potassic carbonate mixed with finely divided carbon. Five parts of tartar should be used in the place of one of charcoal.[Pg 22]
Anthracite or Culm is a kind of coal containing 90 per cent. or more of carbon. It gives off no inflammable gas. It is denser, and takes longer in burning, than charcoal. Its reducing effect is little inferior to that of charcoal. Almost any organic substance can be used as a reducing agent, but it is well not to select one which melts, swells up, or gives off much water and gas when heated in the furnace.
Potassic Cyanide is an easily fusible and somewhat volatile salt, which, when fused, readily removes oxygen and sulphur from metallic compounds, and forms potassic cyanate or sulphocyanate as the case may be. Commercial samples vary much in purity; some contain less than 50 per cent. of the salt. For assaying, only the better qualities should be used.
Iron is a de-sulphurising rather than a de-oxidising agent. Iron is used in the form of rods, 1/2-inch in diameter, or of nails, or of hoop iron. In the last case it should be thin enough to be bent without difficulty. Wrought iron crucibles are very useful in the processes required for making galena assays.
The chief oxidising agents (which are also de-sulphurisers) are the following:—
Nitre, or Potassic Nitrate.—This salt fuses very easily to a watery liquid. It oxidises most combustible substances with deflagration, and thereby converts sulphides into sulphates, arsenides into arsenates, and most metals into oxides. In the presence of strong bases, such as soda, the whole of the sulphur is fully oxidised; but in many cases some arsenic is apt to escape, and to give rise to a peculiar garlic-like odour. The sulphates of soda and potash are thus formed, and float as a watery liquid on the surface of the slag.
Red lead is an oxide of lead. About one-quarter of its oxygen is very loosely held, and, hence, is available for oxidising purposes, without any separation of metallic lead. The rest of the oxygen is also available; but for each part of oxygen given off, about 13 parts of metallic lead are deposited. In silver assays this power of readily giving up oxygen is made use of. The residual oxide (litharge) acts as a flux.
Hot air is the oxidising agent in roasting operations. The sulphur and arsenic of such minerals as mispickel and pyrites are oxidised by the hot air and pass off as sulphur dioxide and "white arsenic." The metals generally remain in the form of oxide, mixed with more or less sulphate and arsenate. The residue may remain as a powdery substance (a calx), in which case the process of roasting is termed calcination; or it may be a pasty mass or liquid. In the calcination of somewhat fusible minerals, the roasting should be done at a low temperature to avoid clotting;[Pg 23] arsenic and sulphur being with difficulty burnt off from the clotted mineral. A low temperature, however, favours the formation of sulphates; and these (if not removed) would reappear in a subsequent reduction as sulphides. These sulphates may be decomposed by a higher temperature towards the end of the operation; their removal is rendered more certain by rubbing up the calx with some culm and re-roasting, or by strongly heating the calx after the addition of solid ammonic carbonate. In roasting operations, as large a surface of the substance as possible should be exposed to the air. If done in a crucible, the crucible should be of the Cornish type, short and open, not long and narrow. For calcinations, roasting dishes are useful: these are broad and shallow, not unlike saucers, but unglazed. In those cases in which the products of the roasting are liquid at the temperature used, a scorifier (fig. 38) is suitable if it is desired to keep the liquid; but if the liquid is best drained off as quickly as it is formed, a cupel (fig. 5) should be used.
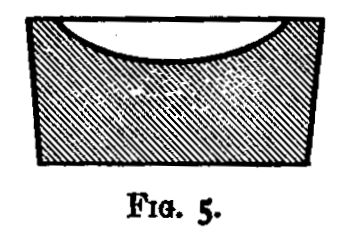
A scorifier is essentially a roasting dish sufficiently thick to resist, for a time, the corrosive action of the fused metallic oxides it is to contain. The essential property of a cupel is, that it is sufficiently porous to allow the fused oxide to drain into it as fast as it is formed. It should be large enough to absorb the whole of the liquid; and of course must be made of a material upon which the liquid has no corrosive action. Cupels do not bear transport well; hence the assayer generally has to make them, or to supervise their making. A quantity of bone ash is carefully mixed with water so that no lumps are formed, and the mixture is then worked up by rubbing between the hands. The bone ash is sufficiently wet when its cohesion is such that it can be pressed into a lump, and yet be easily crumbled into powder. Cupel moulds should be purchased. They are generally made of turned iron or brass. They consist of three parts (1) a hollow cylinder; (2) a disc of metal; and (3) a piston for compressing the bone ash and shaping the top of the cupel. The disc forms a false bottom for the cylinder. This is put in its place, and the cylinder filled (or nearly so) with the moistened bone ash. The bone ash is then pressed into shape with the piston, and the cupel finished with the help of three or four smart blows from a mallet. Before removing the piston, turn it half-way round upon its axis so as to loosen and smooth the face of the cupel. The cupel is got out by pressing up[Pg 24] the disc of metal forming the false bottom; the removal is more easily effected if the mould is somewhat conical, instead of cylindrical, in form. The cupels are put in a warm place to dry for two or three days. A conveniently sized cupel is 1-1/4 inches in diameter and about 3/4 inch high. The cavity of the cupel is about 1/4 inch deep, and something of the shape shown in fig. 5.
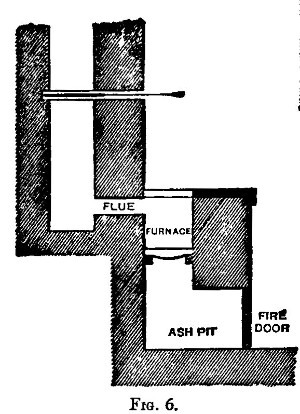
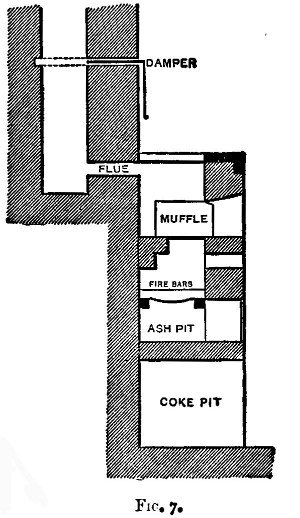
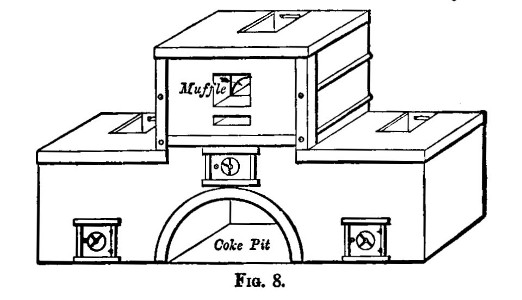
There are two kinds of furnaces required, the "wind" and "muffle" furnaces. These are built of brick, fire-brick, of course, being used for the lining. They are connected with a chimney that will provide a good draught. Figure 6 shows a section of the wind furnace, fig. 7 a section of the muffle furnace, and fig. 8 a general view of a group comprising a muffle and two wind furnaces suitable for general work. When in operation, the furnaces are covered with iron-bound tiles. The opening under the door of the muffle is closed with a loosely fitting brick. The floor of the muffle is protected with a layer of bone-ash, which absorbs any oxide of lead that may be accidentally spilt. The fire bars should be easily removable.
Few tools are wanted; the most important are some cast-iron moulds, tongs (fig. 9), stirrers for calcining (fig. 10), and light tongs of a special form for handling scorifiers and cupels (see Silver).

The coke used should be of good quality; the formation of a fused ash (clinker), in any quantity, causes ceaseless trouble, and requires frequent removal. The coke should be broken into lumps of a uniform size (about 2 in. across) before being brought into the office. The furnace should be well packed by stirring, raising the coke and not ramming it, and it should be uniformly heated, not hot below and cold above. In lighting a furnace, a start is made with wood and charcoal, this readily ignites and sets fire to the coke, which of itself does not kindle easily.
In commencing work, add (if necessary) fresh coke, and mix well; make hollows, and into these put old crucibles; pack around with coke, so that the surface shall be concave, sloping upwards from the mouths of the crucibles to the sides of the furnace; close the furnace, and, when uniformly heated, substitute for the empty crucibles those which contain the assays. It is rarely advisable to have a very hot fire at first, because with a gradual heat the gases and steam quietly escape through the unfused mass, while with too strong a heat these might make some of the matter in the crucible overflow. Moreover, if the heat should be too strong at first, the flux might melt and run to the bottom of the crucible, leaving the quartz, &c., as a pasty mass above; with a gentler heat combination is completed, and the[Pg 26] subsequent fiercer heat simply melts the fusible compound into homogeneous slag.
The fused material may be left in the crucible and separated from it by breaking when cold. It is generally more convenient to pour it into cast-iron moulds. These moulds should be dry and smooth. They act best when warmed and oiled or black-leaded.
Air entering through the fire-bars of a furnace and coming in contact with hot coke combines with it, forming a very hot mixture of carbonic acid and nitrogen; this ascending, comes in contact with more coke, and the carbonic acid is reduced to carbonic oxide; at the top of the furnace, or in the flue, the carbonic oxide meeting fresh air, combines with the oxygen therein and re-forms carbonic acid. In the first and third of these reactions, much heat is evolved; in the second, the furnace is cooled a little. It must always be remembered, that the carbonic oxide of the furnace gases is a reducing agent. When these gases are likely to exert a prejudicial effect, and a strongly oxidising atmosphere is required, the work is best done in a muffle.
[3] There is an exception to this, as when the slag is liable to be acted on when exposed to the air and to the gases of the furnace. In this case a layer of fused common salt floating on the slag, so as to protect it from the air and furnace gases, is a distinct advantage.
In dry assays the metal is almost always separated and weighed as metal; in wet gravimetric assays the metal is more usually weighed in the form of a definite compound of known composition. The general methods of working resemble those of ordinary chemical analysis, and their successful working is greatly helped by a knowledge of, at any rate, those compounds of the metal which enable it to be separated, and of those which are the most convenient forms in which it can be weighed. But the work of the assayer differs from that of the analyst, inasmuch as the bulk of his estimations are made upon material of practically the same kind, varying only in richness; consequently in assaying, it is possible (and necessary) to work on such a definite plan as will involve the least amount of labour in weighing and calculating.
The assayer connected with mining has generally two classes of material to deal with: those comparatively rich and those comparatively poor. For example, silver in bullion and in ores; copper precipitates or regulus, and copper ores and slags; and "black tin" and tin ores. He is only occasionally called on to assay the intermediate products. It is indispensable that he should have an approximate knowledge of the substance to be determined. With new ores this information is best got by a qualitative testing. Knowing that only certain bodies are present, it is evident that the number of separations can be reduced, and that simple methods can be devised for arriving at the results sought for. The best method is that which involves the least number of separations. The reactions must be sharp and complete, and yet not be liable to error under varying conditions.
To bring the richer and poorer materials under the same conditions for the assay, a small weight, say 1 gram of the richer, and a larger weight (5 or 10 grams) of the poorer, substance is weighed up. A method is then adopted which will concentrate the whole of the metal (either during or after solution) in a product which need not necessarily be pure. The work on this product is comparatively easy. In separating small quantities of a substance from a large bulk of impurities, the group separations must not[Pg 28] as a rule be too much relied on. Very large precipitates carry down small quantities of bodies not belonging to the group, more especially when there is a tendency to form weak double compounds. The re-dissolving and re-precipitating of bulky precipitates should be avoided.
When a large number of assays of the same kind have to be carried out, a plan something like the following is adopted:—The samples, after having been dried, are placed in order on a table at the left hand of the assayer. He takes the first, marks it with a number, samples and weighs up the quantity required, and transfers it to a flask, which is similarly marked. As the weighings are finished, the samples are placed in the same order on his right hand. The assistant takes the flasks in batches of four or five at a time to the fume cupboard, where he adds a measured quantity of acid. When solution has been effected, dilution with a measured volume is generally necessary. The assayer sees to this and (whilst the funnels and filters are being prepared) makes any separation that is necessary. The filters are arranged in order on a rack (fig. 11), and need not be marked unless the precipitates or residues have subsequently to be dried. The filters are washed with hot water, and if the filtrates are wanted flasks are placed beneath, if not, the solution is drained off down the sink. Precipitation or reduction (or whatever it may be) is now made; the assistant filters the prepared samples, one at a time, whilst the assayer is engaged with the others. The same style of work is continued until the assays are completed. If one should be spoiled, it is better to allow it to stand over for assaying along with the next batch. If one filters slowly or is in any way less forward than the rest, it may lessen the accuracy of the other assays, owing to oxidation, &c., it should, therefore, be put on one side. The assays are dealt with in batches of ten or twenty, so that a large quantity of work can be quickly finished.
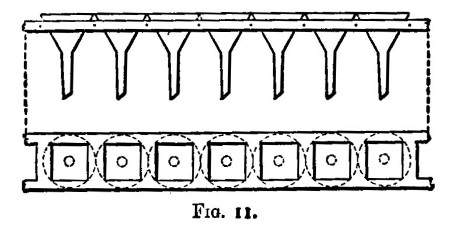
When the assays are finished, it is the duty of the assistant to clean the apparatus (with reagents, if necessary), and to put the[Pg 29] vessels in the place set apart for them. Flasks are best kept inverted on a rack, so that they may be dry and clean by the next morning. Berlin crucibles must be cleaned and ignited.
The amount of apparatus employed should be as little as is feasible. The assay should be carried out as much as possible in the same flask. The bench must be clean, and altogether free from apparatus not in actual use. Crucibles and dishes in which weighings are made should be marked with numbers or letters; and their weights recorded, together with the date of weighing, in a small ledger, which is kept in the drawer of the balance. By this means a record of the "wear" of each piece of apparatus is obtained, and, what is more important, much weighing is saved, and increased confidence is gained. The weight of each piece of apparatus need not be taken daily. It will be seen from the record in the book and a knowledge of the use it has been put to how often a checking of the weight is necessary. The entries are made in black lead as follows:—
| Dish, A. | Feb. 3 | 9.4210 grams. |
| 5 | 9.4225 | |
| 6 | 9.4230 | |
| 7 | 9.4200 |
Platinum vessels and apparatus lose, and porcelain ones slightly gain, weight with continued use.
The special details of the work is given under each assay; certain general instructions will be given here.
Solution.—It is not always necessary to get the whole of the mineral in solution, provided the body sought for is either completely dissolved or altogether left in the residue. It is often only by a qualitative examination of the solution (or residue, as the case may be) that the assayer can satisfy himself that it is free from the substance sought. But previous experience with the same kind of ore will show to what extent this testing is necessary.
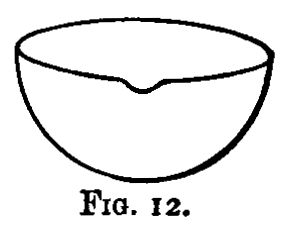
Solution is generally best effected in flasks; but where the resulting liquid has afterwards to be evaporated to dryness and ignited, evaporating dishes (fig. 12) are used. With them clock glasses are used as covers during solution to avoid loss through effervescence. Evaporating dishes are also best when an insoluble residue has to be collected, since it is difficult to wash out most residues from a flask. Bumping occurs less frequently in dishes than in flasks.
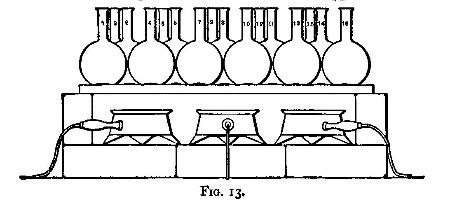
After the addition of the acid, and mixing by agitation, the vessel containing the substance is heated. This is best done on[Pg 30] the "hot plate" (fig. 13). This consists of a slab of cast iron about half or three-quarters of an inch thick, supported on loose fire bricks, and heated by two or three ring burners (figs. 14 and 15). The burners are connected to the gas supply by means of lead tubing, to which they are soldered. Flasks and dishes after being put on the plate are not further handled until solution is complete or the evaporation is carried to dryness. The hot plate is contained in a cupboard so as to be out of the reach of cold draughts.
The action of the acids and other solvents is described in the chapter on Reagents.
Precipitation.—In precipitating add sufficient of the reagent to complete the reaction. The student must be on his guard against adding a very large excess, which is the commoner error. In some reactions the finishing point is obvious enough; either no more precipitate is formed, or a precipitate is completely dissolved, or some well-marked colour or odour is developed or removed.
In those cases in which there is no such indication, theoretical considerations should keep the use of reagents within reasonable limits. The solutions of the reagents (see Reagents) are generally of five or ten per cent. strength. A small excess over that demanded by theory should be sufficient.[Pg 31]
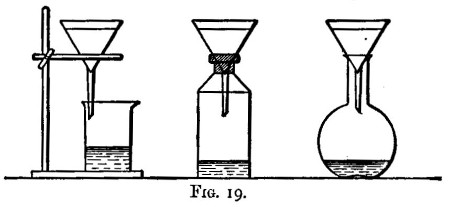
Filtration.—Solutions are best filtered hot whenever the assay allows of this being so done. The precipitate should be allowed to settle, and the clear liquid decanted on the filter with the aid of a glass rod if necessary. The filter-paper must not be too large, but at the same time it must not be overloaded with the precipitate. There should be ample room for washing. For general use three sizes of filter-paper are sufficient. Common quick filtering-paper (English) is best for most work in assaying. The specially prepared paper (Swedish or Rhenish) is used for collecting those precipitates which have to be weighed. The papers are folded as shown in fig. 16, and should not project above the funnel. The filter-paper works better if damped with hot water. In special cases filtering is hastened by means of an air-pump. The apparatus used consists of a water-jet (fig. 17), which is connected with the tap, as also with a bottle fitted as shown in fig. 18. The pump draws the air out from the bottle, and atmospheric pressure forces the liquid through the filter-paper. The bottom of the funnel is provided with a platinum cone, which supports the filter-paper, and prevents its breaking. The pump is only used in exceptional cases; nearly all the filtrations required by the assayer can be made without it. The usual methods of supporting the funnel during filtration are shown in fig. 19. Where the filtrate is not wanted, pickle bottles make convenient supports. After the precipitate has been thrown on the filter, it is washed. In washing, several washings with a small quantity of water are more effective than a few with a[Pg 32] larger quantity of that fluid. The upper edge of the filter-paper is specially liable to escape complete washing. Excessive washing must be avoided; the point at which the washing is complete is found by collecting a little of the filtrate and testing it. The precipitate is removed from the filter-paper for further treatment by opening out the paper and by washing the precipitate with a jet of water from a wash-bottle into a beaker, or back through the funnel into the flask. In some cases, when the precipitate has to be dissolved in anything in which it is readily soluble, solution is effected in the filter itself allowing the liquid to run through as it is formed.
Drying and Igniting.—Precipitates, as a rule, require drying before being ignited. With small precipitates the filter-paper may be opened out, and placed on a warm asbestos slab till dry; or the funnel and the filter with the precipitate is placed in a warm place, and supported by any convenient means. The heat must never be sufficient to char the paper. Some precipitates must be dried at a temperature not higher than 100°C. These are placed in the water-oven (fig. 20), and, when apparently dry, they are taken from the funnel, placed between glasses, and then left in the oven till they cease to lose weight. Such precipitates are collected on tared filters. Those precipitates which will stand a higher temperature are dried in the hot-air oven at a temperature of from 120° to 150°. The drying is continued until they appear to be free from moisture, and until the precipitate ceases to adhere to the filter. In drying sulphides the heat must not be raised to the melting point of sulphur, since, if there is any free sulphur present, it fuses and filters through.
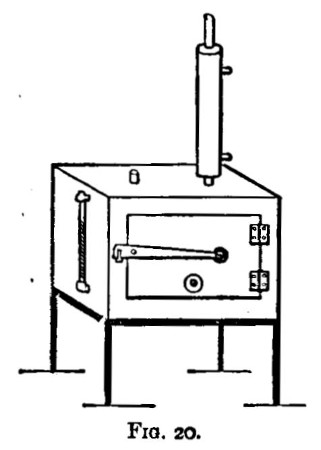
The precipitate, having been dried, is transferred to a watch-glass. The filter-paper is opened out over a sheet of note-paper, and, with a camel-hair brush, the precipitate is gently brought into the glass. Most precipitates come away easily, and the transfer can be made without apparent loss. The watch-glass is covered by the funnel, and the filter-paper (folded into a quadrant) held by the tweezers and set fire to with the flame of a Bunsen burner. It is allowed to burn over the crucible, into which the black bulky ash is allowed to drop, and two or three drops of nitric acid are then added. The crucible is placed on a[Pg 33] pipe-stem triangle (fig. 21), supported on a tripod. It is at first heated gently with a Bunsen burner, and afterwards more strongly, until the residue is free from carbon. It is cooled, and treated with any acid necessary to convert the small amount of precipitate into the state in which it is to be weighed; heated again, and cooled. The main precipitate is transferred to the crucible, and the heating repeated very gently at first, but more strongly towards the end of the operation. It is next placed in the muffle, and, after two or three minutes at a red heat, it is removed and allowed to cool in the desiccator before weighing. This is for bodies that will bear a red heat; for those compounds that require a lower temperature the heating in the muffle is omitted. The muffle used for this purpose must not be used at the same time for cupelling; a gas muffle (fig. 22), such as one of Fletcher's, is best. A desiccator (fig. 23) is an air-tight vessel which prevents access of moisture, &c., to the substance. Usually the air in it is kept dry by means of a basin containing sulphuric acid.
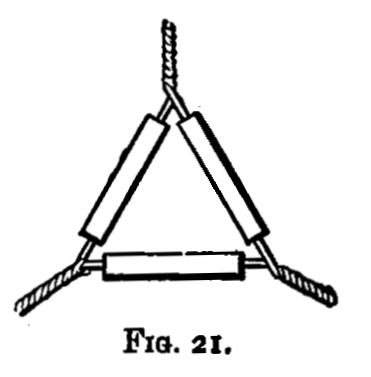
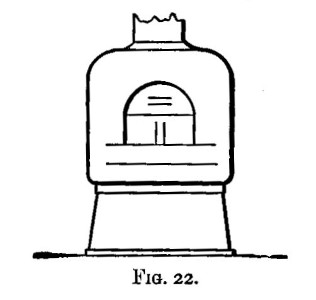
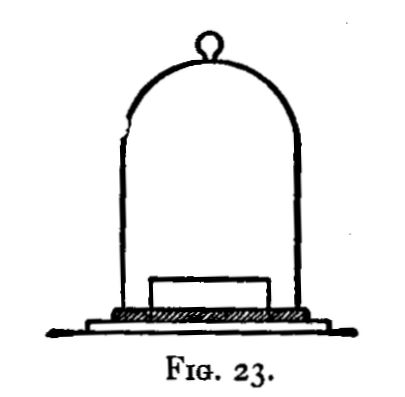
The crucible is removed from the muffle with the tongs and carried to the desiccator. It is best, in an office, to have a large desiccator permanently fixed alongside the balance, into which all substances may be put before being weighed. The substance is removed from the bench or muffle in the small hand apparatus generally sold, and carried to the balance room to be transferred to the large desiccator, where it is allowed to become thoroughly cold before being weighed. Twenty minutes is generally the time allowed after ignition before it is advisable to weigh. Bodies allowed to cool in the air after they have been ignited will absorb moisture, and hot bodies placed in the balance-pan will disturb the equilibrium and show false results. Compounds that absorb moisture must be weighed quickly; they should, therefore, be[Pg 34] weighed in covered vessels. Such compounds are detected by their continually-increasing weight. They should be ignited and weighed again in a well-covered dish.
Substances that have been washed with alcohol, ether, or any readily volatile liquid are dried in the water oven. They quickly dry if there is no water present, and are generally fit for weighing in less than one hour. Sometimes drying for a few minutes only will be sufficient.
The weight of the crucible and precipitate having been obtained, the weight of the crucible and ash is deducted; for example—
| Crucible and precipitate | 10.183 | grams. |
| Crucible and ash | 9.527 | " |
| ——— | ||
| 0.656 | " |
The weight of the ash is best added to that of the crucible. The amount of ash in filter-papers must not be neglected, although papers are now made almost free from ash, and the amount to be deducted is found by taking eight or ten papers and burning them until they become white, and then weighing the ash. The amount varies from 0.004 to 0.0005 gram for different papers. Having determined the ash, place in the balance-drawer three of the filter-papers pinned together, with the weights marked on them in the way shown in fig. 24, so as to be readily seen when there is occasion to refer to them.
It must be remembered that the determination of small quantities of substances generally involves the use of reagents which are often contaminated, as an impurity, with the body sought for. Thus, in assaying silver, the oxide of lead or metallic lead used is rarely free from silver; and in the case of arsenic, the acids, zinc or ferric chloride are sure to contain arsenic. The same observation applies to the precipitation of lead by zinc, &c. The errors caused by these impurities are more marked in the determination of material having small quantities of metal than in that of ores which contain larger quantities. Errors of this kind are counteracted or neutralised by "blank" or "blind" determinations. These consist in carrying out by the side of and during the assay a duplicate experiment with the reagents only, which are thereby subjected to the same processes of solution, evaporation, filtration, &c. The final result thus obtained is deducted from that given by the assay, the difference gives the corrected result. In some cases, where it is desired or necessary to have a tangible residue or precipitate, some pure inert material is added.
These have been already described as those in which the results are got by measuring, either—(1) the volume of a reagent required to complete some reaction, or (2) the volume of the resulting product. For example, if a permanganate of potash solution be added to a solution containing a weighed amount of iron, dissolved in sulphuric acid, the strong colour of the permanganate of potash will be removed until a certain quantity of it has been added. Repeating the experiment, it will be found that the same amount of iron decolorises the same volume of the permanganate solution within certain narrow limits of variation, known as "error of experiment." This error is due to variation in the method of working and to slight differences in the weighings and measurings; it is present in all experimental methods, although the limits of variation are wider in some than in others. Apart from this error of experiment, however, it is certain that a given volume of the permanganate of potash solution corresponds to a definite weight of iron, so that if either is known the other may be calculated. Similarly, if a known weight of zinc (or of carbonate of lime) be dissolved in hydrochloric acid, a gas will be given off which can be measured, and so long as the conditions of the experiment do not vary, the same weight of zinc (or of carbonate of lime) gives off the same volume of gas. The weight of the one can be determined from the volume of the other.
Or, again, the quantity of some substances may be measured by the colour of their solutions, on the principle that, other things being equal, the colour of a solution depends upon the quantity of colouring matter present. So that if two solutions of the same substance are equally coloured they are of equal strength. In this way an unknown may be compared with a known strength, and a fairly accurate determination may be made. These three illustrations serve as types of the three chief classes of volumetric assays—titrometric, gasometric, and colorimetric.
Titrometric Assays.—Within the limits of the error of experiment, a definite volume of a solution or gas represents a certain weight of metal or other substance, hence the exact weight may be[Pg 36] determined by experiment. The error of experiment may be reduced to insignificant dimensions by repeating the experiment, and taking the mean of three or four determinations. This will at the same time show the amount of variation. Thus, if 0.5 gram of iron were dissolved and found to require 50.3 cubic centimetres of the solution of permanganate of potash, and if on repeating, 50.4, 50.2, and 50.3 c.c. were required, the experimenter would be justified in saying that 50.3 c.c. of the permanganate solution represent 0.5 gram of iron, and that his results were good within 0.2 c.c. of the permanganate solution. So that if in an unknown solution of iron, 50.5 c.c. of the permanganate solution were used up, he could state with confidence that it contained a little more than 0.5 gram of iron. With a larger experience the confidence would increase, and with practice the experimental error will diminish.
But supposing that the unknown solution required, say, 100.5 instead of 50.5 c.c., he would not be justified in saying that, since 50.3 c.c. are equivalent to 0.5 gram, 100.6 c.c. are equivalent to twice that amount; and that, consequently, the unknown solution contained a little less than 1 gram of iron; or, at least, he could not say it except he (or some one else) had determined it by experiment. But if on dissolving 1 gram of iron, he found it to require 100.6 c.c. of the solution, and in another experiment with 0.8 gram of iron that 80.5 c.c. of the solution were required, he would be justified in stating that the volume of solution required is proportional to the quantity of metal present. There are a large number of volumetric assays of which this is true, but that it is true in any particular case can only be proved by experiment. Even where true it is well not to rest too much weight upon it, and in all cases the quantity of metal taken, to determine the strength of the solution used, should not differ widely from that present in the assay. There are certain terms which should be explained here. When the solution of a reagent is applied under such conditions that the volume added can be correctly determined, the operation is called "titrating," the solution of the reagent used the "standard solution," and the process of determining the strength of the standard solution is "standardising." The "standard" is the quantity of metal equivalent to 100 c.c. of the standard solution.
Standard Solutions.—In making these the salt is accurately weighed and transferred to a litre flask, or to the graduated cylinder, and dissolved. The method of dissolving it varies in special cases, and instructions for these will be found under the respective assays. Generally it is dissolved in a small quantity of liquid, and then diluted to the mark. For those substances that require the aid of heat, the solution is made in a pint flask, cooled,[Pg 37] and transferred; after which the flask is well washed out. After dilution, the liquids in the measuring vessel must be thoroughly mixed by shaking. This is more easily and better done in the cylinder than in the litre flask. The solution is next transferred to a dry "Winchester" bottle and labelled. The label may be rendered permanent by waxing it.
Standard solutions should not be kept in a place exposed to direct sunlight. Oxidising and reducing solutions, such as those of permanganate of potash, ferrous sulphate, iodine, hyposulphite of soda, &c., gradually weaken in strength; the solutions of other salts are more stable; while those of potassium bichromate and baric chloride are almost permanent. Solutions of potassium permanganate may be kept for a month or so without much change. The solutions of hyposulphite of soda and of iodine should be examined weekly. Ferrous sulphate solutions, if acidulated with sulphuric acid, may be depended on for two or three weeks without fresh standardising. Before filling the burette, the "Winchester" bottle should be well shaken and a portion of about 50 or 100 c.c. poured into a dry beaker or test-glass. Besides the standard solutions, which are required for titrating an assay, permanent solutions of the metal or acid of equivalent strength are very useful. When the finishing point of a titration has been overstepped (i.e., the assay has been "overdone"), a measured volume, say 5 or 10 c.c., of a solution containing the same metal may be added. The titration can then be continued, but more cautiously, and the value in "c.c." for the quantity added be deducted from the final reading.
Standardising.—Suppose the object is to standardise a solution of permanganate similar to that referred to above. A convenient quantity of iron (say 0.5 gram) would be weighed out, dissolved in dilute sulphuric acid, and the solution titrated. Suppose 49.6 c.c. of the permanganate solution are required, then
49.6 : 0.5 :: 100 : x
x = 1.008 gram.
This result, 1.008 gram, is the "standard." When a gas is measured, the standard may be calculated in the same way. For example: with 0.224 gram of zinc, 75.8 c.c. of gas were obtained. Then the quantity of zinc equivalent to 100 c.c. of the gas is got by the proportion.
75.8 : 0.224 :: 100 : x
x = 0.2955 gram.
Using the term "standard" in this sense, the following rules hold good:[Pg 38]—
To find the weight of metal in a given substance:—Multiply the standard by the number of c.c. used and divide by 100. For example: a piece of zinc was dissolved and the gas evolved measured 73.9 c.c. Then by the rule, 0.2955 × 73.9 / 100 should give the weight of the piece of zinc. This gives 0.2184 gram.
To find the percentage of metal in a given substance:—Multiply the standard by the number of c.c. used and divide by the weight of substance taken. For example: if 2 grams of a mineral were taken, and if on titrating with the permanganate solution (standard 1.008) 60.4 c.c. were required, then (1.008×60.4)/2 = 30.44. This is the percentage.
If the standard is exactly 1 gram, and 1 gram of ore is always taken, these calculations become very simple. The "c.c." used give at once the percentage, or divided by 100 give the weight of metal.
If it is desired to have a solution with a standard exactly 1.0 gram, it is best first to make one rather stronger than this, and then to standardise carefully. Divide 1000 by the standard thus obtained and the result will be the number of c.c. which must be taken and be diluted with water to 1 litre. For example: suppose the standard is 1.008, then 1000/1.008 gives 992, and if 992 c.c. be taken and diluted with water to 1000 c.c. a solution of the desired strength will be obtained. The standard of this should be confirmed. A simpler calculation for the same purpose is to multiply the standard by 1000; this will give the number of c.c. to which 1 litre of the solution should be diluted. In the above example a litre should be diluted to 1008 c.c.
It has been assumed in these rules that the titration has yielded proportional results; but these are not always obtained. There can be no doubt that in any actual re-action the proportion between any two re-agents is a fixed one, and that if we double one of these then exactly twice as much of the other will enter into the re-action; but in the working it may very well be that no re-action at all will take place until after a certain excess of one or of both of the re-agents is present. In titrating lead with a chromate of potash solution, for example, it is possible that at the end of the titration a small quantity of the lead may remain unacted on; and it is certain that a small excess of the chromate is present in the solution. So, too, in precipitating a solution of silver with a standard solution of common salt, a point is reached at which a small quantity of each remains in solution; a further addition either of silver or of salt will cause a precipitate, and a similar phenomenon has been observed in precipitating a hydrochloric acid solution of a sulphate with[Pg 39] baric chloride. The excess of one or other of the re-agents may be large or small; or, in some cases, they may neutralise each other. Considerations like these emphasise the necessity for uniformity in the mode of working. Whether a process yields proportional results, or not, will be seen from a series of standardisings. Having obtained these, the results should be arranged as in the table, placing the quantities of metal used in the order of weight in the first column, the volumes measured in the second, and the standards calculated in the third. If the results are proportional, these standards will vary more or less, according to the delicacy of the process, but there will be no apparent order in the variation. The average of the standards should then be taken.
| Weight. | Volume found. | Standard |
| 0.2160 gram | 72.9 c.c. | 0.2963 |
| 0.2185 " | 73.9 " | 0.2957 |
| 0.2365 " | 79.9 " | 0.2959 |
| 0.2440 " | 82.3 " | 0.2964 |
| 0.2555 " | 85.9 " | 0.2974 |
Any inclination that may be felt for obtaining an appearance of greater accuracy by ignoring the last result must be resisted. For, although it would make no practical difference whether the mean standard is taken as 0.2961 or 0.2963, it is well not to ignore the possibility that an error of 0.4 c.c. may arise. A result should only be ignored when the cause of its variation is known.
In this series the results are proportional, but the range of weights (0.216-0.2555 gram) is small. All processes yield fairly proportional results if the quantities vary within narrow limits.
As to results which are not proportional, it is best to take some imaginary examples, and then to apply the lesson to an actual one. A series of titrations of a copper solution by means of a solution of potassic cyanide gave the following results:—
| Copper taken. | Cyanide used. | Standard. |
| 0.1 gram | 11.9 c.c. | 0.8403 |
| 0.2 " | 23.7 " | 0.8438 |
| 0.3 " | 35.6 " | 0.8426 |
| 0.4 " | 47.6 " | 0.8403 |
These are proportional, but by using a larger quantity of acid and ammonia in the work preliminary to titration, we might have had[Pg 40] to use 1 c.c. of cyanide solution more in each case before the finishing point was reached. The results would then have been:
| Copper taken. | Cyanide used. | Standard. |
| 0.1 gram | 12.9 c.c. | 0.7752 |
| 0.2 " | 24.7 " | 0.8097 |
| 0.3 " | 36.6 " | 0.8191 |
| 0.4 " | 48.6 " | 0.8230 |
It will be noted that the value of the standard increases with the weight of metal used; and calculations from the mean standard will be incorrect.
By subtracting the lowest standardising from the highest, a third result is got free from any error common to the other two; thus:—
| 0.4 | gram | = | 48.6 | c.c. | "cyanide." |
| 0.1 | " | = | 12.9 | " | " |
| —— | —— | ||||
| 0.3 | " | = | 35.7 | " | " |
And the standard calculated from this corrected result is 0.8404. Further, if 0.3 gram requires 35.7 c.c., then 0.1 gram should require 11.9 c.c., or 1.0 c.c. less than that actually found.
We may therefore use the following rules for working processes which do not yield proportional results. Make a series of two or three titrations, using very different quantities of metal in each. Subtract the lowest of these from the highest, and calculate the standard with the remainder. Calculate the volume required by this standard in any case, and find the excess or deficit, as the case may be. If an excess, subtract it from the result of each titration; if a deficit, add it; and use the standard in the usual way. The following table shows an actual example:—
| Chalk taken. | Gas obtained. | Standard. |
| 0.0873 gram | 17.8 c.c. | 0.4904 |
| 0.1305 " | 27.3 " | 0.4780 |
| 0.1690 " | 35.8 " | 0.4721 |
| 0.1905 " | 40.4 " | 0.4715 |
| 0.2460 " | 52.5 " | 0.4686 |
| 0.3000 " | 64.0 " | 0.4687 |
It will be seen that the standard decreases as the quantity of chalk increases; this points to a deficiency in the quantity of gas evolved.[Pg 41]
Then
| 0.3000 | = | 64.0 | c.c. |
| 0.0873 | = | 17.8 | " |
| ——— | = | —— | |
| 0.2127 | = | 46.2 | " |
and 0.2127×100/46.2 = 0.4604. Then, multiplying the weight of chalk taken by 100, and dividing by 0.4604, we get the calculated results of the following table:—
| Chalk taken. | Gas found. | Gas calculated. | Difference. |
| 0.0873 gram | 17.8 c.c. | 18.9 c.c. | -1.1 c.c. |
| 0.1305 " | 27.3 " | 28.3 " | -1.0 " |
| 0.1690 " | 35.8 " | 36.7 " | -0.9 " |
| 0.1905 " | 40.4 " | 41.4 " | -1.0 " |
| 0.2460 " | 52.5 " | 53.4 " | -0.9 " |
| 0.3000 " | 64.0 " | 65.1 " | -1.1 " |
By adding 1 c.c. to the quantity of gas obtained, and taking 0.4604 as the standard, the calculated results will agree with those found with a variation of 0.1 c.c. When a large number of assays of the same kind are being made, this method of calculation is convenient; when, however, only one or two determinations are in question, it is easier to make a couple of standardisings, taking quantities as nearly as possible the same as those present in the assays.
Sometimes it is necessary to draw up a table which will show, without calculation, the weight of substance equivalent to a given volume of gas or of solution. The substance used for standardising should be, whenever possible, a pure sample of the substance to be determined—that is, for copper assays pure copper should be used, for iron assays pure iron, and so on; but when this cannot be got an impure substance may be used, provided it contains a known percentage of the metal, and that the impurities present are not such as will interfere with the accuracy of the assay. Including compounds with these, the standard may be calculated by multiplying the standard got in the usual way, by the percentage of metal in the compound or impure substance, and dividing by 100. If, for example, the standard 1.008 gram was obtained by using a sample of iron containing 99.7 per cent. of metal, the corrected standard would be 1.008×99.7/100 = 1.005.
In volumetric analysis the change brought about must be one in which the end of the reaction is rendered prominent either by a change of colour or by the presence or absence of a precipitate. If the end of the reaction or finishing-point is not of itself visible,[Pg 42] then it must be rendered visible by the use of a third reagent called an indicator.
For example, the action of sulphuric acid upon soda results in nothing which makes the action conspicuous; if, however, litmus or phenolphthalein be added the change from blue to red in the first case, or from red to colourless in the second, renders the finishing-point evident. Some indicators cannot be added to the assay solution without spoiling the result; in which case portions of the assay solution must be withdrawn from time to time and tested. This withdrawal of portions of the assay solution, if rashly done, must result in loss; if, however, the solution is not concentrated, and if the portions are only withdrawn towards the end of the titration, the loss is very trifling, and will not show-up on the result. The usual plan adopted is to have a solution of the indicator placed in drops at fairly equal intervals distributed over a clean and dry white porcelain-plate: a drop or two of the solution to be tested is then brought in contact with one of these and the effect noted. Another plan is to have thin blotting-paper, moistened with a solution of the indicator and dried; a drop of the solution to be tested placed on this shows the characteristic change. When the assay solution contains a suspended solid which interferes with the test, a prepared paper covered with an ordinary filter-paper answers very well; a drop of the solution to be tested is placed on the filter-paper, and, sinking through, shows its effect on the paper below.
Except when otherwise stated, all titrations should be made at the ordinary temperature; cooling, if necessary, by holding the flask under the tap. When a titration is directed to be made in a boiling solution, it must be remembered that the standard solution is cold, and that every addition lowers the temperature of the assay.
On running the solution from the burette into the assay, do not let it run down the side of the flask. If a portion of the assay has to be withdrawn for testing, shake the flask to ensure mixing, and then take out a drop with the test-rod; the neglect of these precautions may give a finishing-point too early. This is generally indicated by a sudden finish, in which case on shaking the flask and again testing no reaction is got. Do not remove the drop on the point of the burette with the test-rod; let it remain where it is or drop it into the solution by carefully opening the clip.
Generally the methods of working are as follows:—
(1) When the finishing-point depends on a change of colour in the solution.—Increase the bulk of the assay up to from 100 to 150 c.c. with water. Boil or cool, as the case may be. Run in the standard solution from a burette speedily, until the re-agent appears to have a slower action, and shake or stir all the time.[Pg 43] Then run 1 c.c. or so at a time, still stirring, and finally add drops until the colour change is got.
(2) When an outside-indicator is used.—Pour the standard solution from a burette into the assay until 5 or 6 c.c. from the finishing-point; then run in 1 c.c. at a time (stirring and testing on the plate between each) until the indicator shows the change wanted, and deduct 0.5 c.c. for excess. When greater accuracy is sought for a duplicate assay is made. In this case the standard solution is run in close up to the end, and the operation is finished off with a few drops at a time.
(3) Where the finishing-point depends upon the absence of a precipitate and no outside-indicator is used.—As in the last case, run in the standard solution up to within a few c.c. of the end, then run in 1 c.c. at a time until a precipitate is no longer formed, but here 1.5 c.c. must be deducted for excess, since it is evident that the whole of the last "c.c." must have been, and a portion of the previous one may have been, in excess.
Indirect Titration.—The action of permanganate of potash upon a ferrous solution is one of oxidation, hence it is evident that if any other oxidising agent is present it will count as permanganate. In such a case the titration can be used (indirectly) to estimate the quantity of such oxidising agent, by determining how much less of the permanganate is used. For example, suppose that 1 gram of iron dissolved in sulphuric acid requires 100 c.c. of standard permanganate to fully oxidise it, but that the same amount of iron only requires 35.6 c.c. of the same standard permanganate if it has been previously heated with 0.5 gram of black oxide of manganese. Here it is evident that 0.5 gram of black oxide does the work of 64.4 c.c.[4] of the permanganate solution, and that these quantities are equivalent; moreover, if 64.4 c.c. correspond with 0.5 gram, then 100 c.c. correspond with 0.7764 which is the standard. On theoretical grounds, and by a method of calculation which will be explained further on (under the heading "Calculations from Formulæ"), it can be found that if the standard for iron is 1 gram, that for the black oxide will be 0.7764 gram.
The principles of these indirect titrations become clearer when expressed in a condensed form. Thus, in the example selected, and using the formulæ Fe = Iron, KMnO4 = permanganate of potash, and MnO2 = oxide of manganese, we have:—
(1) 1 gram Fe = 100 c.c. KMnO4
(2) 1 gram Fe = 35.6 c.c. KMnO4 + 0.5 gram MnO2
∴ 100 c.c. KMnO4 = 35.6 c.c. KMnO4 + 0.5 gram MnO2
(100 - 35.6) c.c. KMnO4 = 0.5 gram MnO2
64.4 c.c. KMnO4 = 0.5 gram MnO2
[Pg 44]
The iron does not enter into the calculation if the same quantity is present in the two experiments.
An indirect titration thus requires three determinations, but if more than one assay is to be carried on, two of these need not be repeated. The standard is calculated in the usual way.
Colorimetric Assays.—These are assays in which the colour imparted to a solution by some compound of the metal to be determined is taken advantage of; the depth of colour depending on the quantity of metal present. They are generally used for the determination of such small quantities as are too minute to be weighed. The method of working is as follows:—A measured portion of the assay solution (generally 2/3, 1/2, 1/3, or 1/4 of the whole), coloured by the substance to be estimated, is placed in a white glass cylinder standing on a sheet of white paper or glazed porcelain. Into an exactly similar cylinder is placed the same amount of re-agents, &c., as the portion of the assay solution contains, and then water is added until the solutions are of nearly equal bulk. Next, a standard solution of the metal being estimated is run in from a burette, the mixture being stirred after each addition until the colour approaches that of the assay. The bulk of the two solutions is equalised by adding water. Then more standard solution is added until the tints are very nearly alike. Next, the amount added is read off from the burette, still more is poured in until the colour is slightly darker than that of the assay, and the burette read off again. The mean of the readings is taken, and gives the quantity of metal added. It equals the quantity of metal in the portion of the assay. If this portion was one-half of the whole, multiply by two; if one-third, multiply by three, and so on. When the quantity of metal in very dilute solutions is to be determined, it is sometimes necessary to concentrate the solutions by boiling them down before applying the re-agent which produces the coloured compound. Such concentration does not affect the calculations.
Gasometric Assays.—Gasometric methods are not much used by assayers, and, therefore, those students who wish to study them more fully than the limits of this work will permit, are recommended to consult Winkler and Lunge's text-book on the subject. The methods are without doubt capable of a more extended application. In measuring liquids, ordinary variations of temperature have but little effect, and variations of atmospheric pressure have none at all, whereas with gases it is different. Thus, 100 c.c. of an ordinary aqueous solution would, if heated from 10° C. to 20° C., expand to about 100.15 c.c. 100 c.c. of a gas similarly warmed would expand to about 103.5 c.c., and a fall of one inch in the barometer would have a very similar effect. And in measuring[Pg 45] gases we have not only to take into account variations in volume due to changes in temperature and atmospheric pressure, but also that which is observed when a gas is measured wet and dry. Water gives off vapour at all temperatures, but the amount of vapour is larger as the temperature increases.
By ignoring these considerations, errors of 3 or 4 per cent. are easily made; but, fortunately, the corrections are simple, and it is easy to construct a piece of apparatus by means of which they may be reduced to a simple calculation by the rule of three.
The volume of a gas is, in practice, usually reduced to that which it would be at a temperature of 0° C., when the column of mercury in the barometer is 760 mm. high. But, although convenient, this practice is not always necessary. The only thing required is some way of checking the variations in volume, and of calculating what the corrected volume would be under certain fixed conditions.
Suppose that at the time a series of standardisings is being made, 100 c.c. of air were confined in a graduated tube over moist mercury. These 100 c.c. would vary in volume from day to day, but it would always be true of them that they would measure 100 c.c. under the same conditions as those under which the standardisings were made. If, then, in making an actual assay, 35.4 c.c. of gas were obtained, and the air in the tube measured 105 c.c., we should be justified in saying, that if the conditions had been those of the standardising, the 105 c.c. would have measured 100 c.c., and the 35.4 c.c. would have been 33.7; for 105: 100:: 35.4: 33.7. The rule for using such a piece of apparatus for correcting volumes is:—Multiply the c.c. of gas obtained by 100, and divide by the number of c.c. of air in the apparatus.
If it is desired to calculate the volumes under standard conditions (that is, the gas dry, at 0° C. and 760 mm. barometric pressure) the calculations are easily performed, but the temperature and pressure must be known.
Correction for Moisture.—The "vapour tension" of water has been accurately determined for various temperatures, and it may be looked upon as counteracting the barometric pressure. For example, at 15° C. the vapour tension equals 12.7 millimetres of mercury; if the barometer stood at 750 mm., the correction for moisture would be made by subtracting 12.7 from 750, and taking 737.3 mm. to be the true barometric pressure.
The vapour tensions for temperatures from 0° C. to 20° C. are as follows:[Pg 46]—
| Temp. | Tension. | Temp. | Tension. | Temp. | Tension. |
| 0° | 4.6 mm. | 7° | 7.5 mm. | 14° | 11.9 mm. |
| 1° | 4.9 mm. | 8° | 8.0 mm. | 15° | 12.7 mm. |
| 2° | 5.3 mm. | 9° | 8.6 mm. | 16° | 13.5 mm. |
| 3° | 5.7 mm. | 10° | 9.2 mm. | 17° | 14.4 mm. |
| 4° | 6.1 mm. | 11° | 9.8 mm. | 18° | 15.3 mm. |
| 5° | 6.5 mm. | 12° | 10.5 mm. | 19° | 16.3 mm. |
| 6° | 7.0 mm. | 13° | 11.2 mm. | 20° | 17.4 mm. |
The correction for pressure is:—Multiply the volume by the actual pressure and divide by 760.
The correction for temperature:—Multiply the volume by 273 and divide by the temperature (in degrees Centigrade) added to 273.
For all three corrections the following rules hold good. To reduce to 0° C. and 760 mm. dry.
Volume × 0.3592 × (Pressure-tension)
Corrected volume = ———————————————————
Temperature + 273
To find the volume, which a given volume under standard conditions would assume, if those conditions are altered.
Volume × 2.784 × (Temperature + 273)
Resulting volume = ——————————————————
Pressure - tension
As an example, we will suppose that it is desired to enclose in the apparatus referred to on p. 45, a volume of air, which, when dry (at 0° C. and 760 mm.), shall measure 100 c.c., whilst the actual temperature is 15° C., and the pressure 750 mm.
The second formula is the one to be used, and we get 108.7 c.c.
100 c.c.×2.784×288
Required volume = ———————————
750-12.7
80179.2
= ————
737.3
= 108.7 c.c.
[4] 100-35.6 = 64.4.
Weighing.—The system of weights and measures which we have adopted is the French or metric system; in this the gram (15.43 grains) is the unit of weight; the only other weight frequently referred to is the milligram, which is 0.001, or 1/1000 gram. The unit of volume is the cubic centimetre, which is approximately the volume of 1 gram of water, and which thus bears to the gram the same relation as grain-measures bear to grains. It is usual to write and even pronounce cubic centimetre shortly as c.c., and the only other denomination of volume we shall have occasion to use is the "litre," which measures 1000 c.c., and is roughly 1-3/4 pints.
The weights used are kept in boxes in a definite order, so that the weights on the balance can be counted as well by noting those which are absent from the box as by counting those present on the scale-pan. The weights run 50, 20, 10, 10, 5, 2, 1, 1 and 1 grams, and are formed of brass. The fractions of the gram are generally made of platinum or of aluminium, and are arranged in the following order:—0.5, 0.2, 0.1, 0.1, and 0.05, 0.02, 0.01, 0.01. These may be marked in this way, or they may be marked 500, 200, 100, 100, 50, 20, 10, 10; the 500 meaning 500 milligrams.
Some makers send out weights in the series 50, 20, 20, 10, &c.
Weights of less than 0.01 gram are generally present in a box, but it is much more convenient to work with a rider. This is a piece of wire which in the pan weighs 0.01 gram; it is made in such a form that it will ride on the beam, and its effective weight decreases as it approaches the centre. If the arm of the beam is divided into tenths, then each tenth counting from the centre outward equals 0.001 gram or 1 milligram, and if these tenths be further subdivided the fractions of a milligram are obtained; and these give figures in the fourth place of decimals. A fairly good balance should be sensitive to 0.0001 gram. The weights must never be touched with the fingers, and the forceps for moving them is used for no other purpose. When not in actual use the box is kept closed. The weights must not be allowed to remain on the pan of the balance. The balance-case must not be[Pg 48] open without some reason. It must be fixed level, and, once fixed, must not be needlessly moved. The bench on which it stands should be used for no other purpose, and no one should be allowed to lean upon it.
When using a balance sit directly in front of it. Ordinarily the substance to be weighed is best put on the pan to the user's left; the weights and the rider are then easily manipulated. Powders, &c., should not be weighed directly on the balance; a counterpoised watch-glass or metal scoop (fig. 25) should be used. In some cases it is advisable to use a weighing-bottle. This is a light, well-stoppered bottle (fig. 3) containing the powdered ore. It is first filled and weighed; then some of the substance is carefully poured from it into a beaker or other vessel, and it is weighed again; the difference in the two weighings gives the weight of substance taken. A substance must always be cold when weighed, and large glass vessels should be allowed to stand in the balance-box a little while before being weighed. Always have the balance at rest when putting on or taking off anything from the pans. Put the weights on systematically. In using the rider (except you have a reason to the contrary), put it on at the 5; if this is too much, then try it at the 3; if then the weights are too little, try at the 4, if still not enough, the correct weight must be between the 4 and 5; try half-way between.
It is best to work with the balance vibrating; equilibrium is established when the vibration to the left is the mean of the preceding and succeeding vibrations to the right. For example, if it vibrates 6 divisions to the right on one swing, and 5 divisions on the next, the intermediate vibration to the left should have been 5-1/2.
Note whether the substance increases in weight whilst on the balance. If it does it may be because it was put on warm, and is cooling, or it may be because it is taking up moisture from the air. Substances which take up moisture rapidly should be weighed in clipped watch-glasses or in light-weighing bottles or tubes.
Students, in recording the weights, should first read off those missing from the box, writing down each order of figures as determined; first tens, then units, and so on. Remember that the first four platinum weights give the figures of the first place of decimals, the second four give the second place, and that the third and fourth places are given by the rider. Having taken down the figures, confirm them by reading off the weights as you put them[Pg 49] back into the box. Do not rest a weight on the palm of your hand for convenience in reading the mark upon it. Remember one weight lost from a box spoils the set. Do not take it for granted that the balance is in equilibrium before you start weighing: try it.
Measuring Liquids.—For coarse work, such as measuring acids for dissolving ores, graduated glasses similar to those used by druggists may be used. It is well to have two sizes—a smaller graduated into divisions of 5 c.c. (fig. 26), and a larger with divisions equal to 10 c.c. No measurement of importance should be made in a vessel of this kind, as a slight variation in level causes a serious error.
Graduated flasks must be used when anything has to be made up to a definite bulk, or when a fixed volume has to be collected. If, for example, a certain weight of substance has to be dissolved and diluted to a litre, or if the first 50 c.c. of a distillate has to be collected, a flask should be used. Each flask is graduated for one particular quantity; the most useful sizes are 1000 c.c., 500 c.c., 200 c.c., 100 c.c., and 50 c.c. The mark should be in the narrowest part of the neck, and should be tangential to the curved surface of the liquid when the flask contains the exact volume specified. The level of a curved surface of liquid is at first somewhat difficult to read: the beginner is in doubt whether the surface should be taken at a, b, or c (fig. 27). It is best to take the lowest reading c. In some lights it is difficult to find this; in such cases a piece of white paper or card held behind and a little below, so as to throw light up and against the curved surface, will render it clear. In reading, one should look neither up at nor down upon the surface, but the eye should be on the same level with it. It must be kept in mind that flasks contain the quantity specified, but deliver less than this by the amount remaining in them and damping the sides. If it is desired to transfer the contents say of a 100 c.c. flask to a beaker, it will be necessary to complete the transfer by rinsing out the flask and adding the washings; otherwise there will be a sensible loss. Graduated cylinders (fig. 28) are convenient for preparing standard solutions.
Pipettes and burettes are graduated to deliver the quantities[Pg 50] specified. The principle of the pipette, and the advantages and disadvantages of its various forms, may be understood by considering the first form shown in fig. 29. It is essentially a bulbed tube drawn out to a jet at its lower end, and having on each side of the bulb a mark so placed that when the surface of the liquid falls from the upper to the lower mark the instrument shall deliver exactly 100 c.c. The bore of the jet should be of such a size as will allow the level of the liquid to fall at the rate of about one foot in two minutes. If it runs more quickly than this, an appreciable error arises from the varying amount of liquid remaining, and damping the sides of the bulb. The flow of liquid from a pipette must not be hastened by blowing into it. The lower tube or nose of the pipette should be long enough to reach into the bottle or flask containing the liquid about to be measured. The pipette is filled by sucking at the open end with the mouth; this method of filling renders the use of the instrument dangerous for such liquids as strong acids, ammonia, and such poisonous solutions as that of potassic cyanide. One attempt with a fairly strong solution of ammonia will teach the beginner a very useful lesson. As soon as the liquid rises above the upper mark in the pipette, the mouth is withdrawn, and the pipette quickly closed by pressing the upper aperture with the index finger of the right hand; it is well to have the finger slightly moist, but not damp. The neck of the pipette should be long enough to allow its being firmly grasped by the fingers and thumb of the right hand without inconvenience. The pipette is first held in a vertical position long enough to allow any moisture outside the tube to run down, and then the liquid is allowed to run out to the level of the upper mark; this is easily effected by lessening the pressure. If the finger is wet, the flow will be jerky, and good work impossible. The pipette is next held over the vessel into which the 100 c.c. are to be put, and the liquid allowed to run out. When the bulb is nearly empty, the flow should be checked by replacing the finger, and the liquid allowed to escape slowly until the lower mark is reached. The pipette is then withdrawn; it is in the withdrawing that the disadvantage of this particular form[5] makes itself felt. It must be withdrawn[Pg 51] very steadily, as the slightest shock causes the remaining column of liquid to vibrate, whereby air is drawn in and the liquid is forced out.
This disadvantage is got rid of by making the mouth of the jet the lower limit, or, in other words, allowing the instrument to empty itself. There are two forms of such pipettes; in the one generally recommended in Gay-Lussac's silver assay (the last shown in fig. 29) the nose is replaced by a jet. This is most conveniently filled by stopping the jet with the finger, and allowing the liquid to flow in a fine stream into the neck until the pipette is filled, and then working as just described. The other form is the one in general use; in fact, a long nose to a pipette is so convenient that it may almost be said to be necessary. But the accuracy is slightly diminished; a long narrow tube makes a poor measuring instrument because of the amount of liquid it finally retains. A defect possessed by both forms is the retention of a drop of varying size in the nozzle. Whatever method is adopted for removing this drop must be always adhered to. The most convenient form is the one last described, and the most useful sizes are 100 c.c., 50 c.c., 20 c.c., 10 c.c., and 5 c.c. Ten c.c. pipettes graduated into tenths of a cubic centimetre are very useful: those are best in which the graduation stops short of the bottom.
All measurements should be made at the ordinary temperature; and, before being used, the pipette should be rinsed out with a cubic centimetre or so of the solution to be measured. After using, it should be washed out with water.
Burettes differ mainly from pipettes in having the flow of liquid controlled from below instead of from above. The best form is that known as Mohr's, one kind of which is provided with a glass stopcock, while the other has a piece of india-rubber tube compressed by a clip. The latter cannot be used for solutions of permanganate of potash or of iodine, or of any substance which acts on india-rubber; but in other respects there is little to choose between the two kinds. A burette delivering 100 c.c., and graduated into fifths (i.e., each division = 0.2 c.c.), is a very convenient size. For some kinds of work, 50 c.c. divided into tenths (i.e., each division = 0.1 c.c.) may be selected.
Burettes may be fixed in any convenient stand; they must be vertical and should be so placed that the assayer can read any part of the graduated scale without straining. When not in use, they should be kept full of water. When using a burette, the water must be run out; the burette is next rinsed with some of the solution to be used, and drained; and then it is filled with the solution. Next squeeze the india-rubber tube so as to disentangle air-bubbles and, by smartly opening the clip, allow the tube and[Pg 52] jet to be filled; see that no bubbles of air are left. Then run out cautiously until the level of the liquid in the burette stands at zero. In reading the level with very dark-coloured liquids it is convenient to read from the level a (fig. 27), and, provided it is done in each reading, there is no objection to this. The accuracy of the reading of a burette is sensibly increased by the use of an Erdmann float. This is an elongated bulb, weighted with mercury, and fitting (somewhat loosely) the tube of the burette. It floats in the solution, and is marked with a horizontal line; this line is taken as the level of the liquid. If the burette is filled from the top, the float rises with aggravating slowness, and this is its chief disadvantage. The float must come to rest before any reading is made.
A convenient plan for filling a burette from below is shown in fig. 30. The diagram explains itself. The bottle containing the standard solution is connected with the burette by a syphon arrangement through the glass tube and T-piece. The flow of liquid into the burette is controlled by the clip. When this clip is opened, the burette fills; and when it is closed, the burette is ready for use in the ordinary way.
Measuring Gases.—Lange's nitrometer (fig. 69) is a very convenient instrument for many gasometric methods. It requires the use of a fair quantity of mercury. In fig. 31, there is a representation of a piece of apparatus easily fitted up from the ordinary material of a laboratory. It is one which will serve some useful purposes. It consists of a wide-mouthed bottle fitted (by preference) with a rubber cork. The cork is perforated, and in the perforation is placed a glass tube which communicates with the burette. The burette is connected by a rubber tube and a Y-piece, either with another burette or with a piece of ordinary combustion-tube of about the same size. The wide-mouthed bottle contains either a short test-tube or an ordinary phial with its neck cut off. In working the apparatus the weighed substance is put in the bottle and the re-agent which[Pg 53] is to act on it, in the test-tube; the cork is then inserted. The liquid in the two burettes is next brought to the same level, either by pouring it in at a or running it out at b. The level of the liquid in the apparatus for correcting variation in volume is then read and noted. Next, after seeing that the level of the liquid in the burette has not changed, turn the bottle over on its side so that the re-agent in the test-tube shall be upset into the bottle. Then, as the volume of the gas increases, lower the liquid in the burette by running it out at b, and at the same time keep the level in a half an inch or so lower than that in the burette. When the action has finished bring the liquid in the two vessels to the same level and read off the burette. This part of the work must always be done in the same manner.
The volume corrector for gas analysis is a graduated glass tube of 120 c.c. capacity inverted over a narrow glass cylinder of mercury. It contains 0.2 or 0.3 c.c. of water and a volume of air, which, if dry and under standard conditions, would measure 100 c.c. The actual volume varies from day to day, and is read off at any time by bringing the mercury inside and outside to the same level. This is done by raising or lowering the tube, as may be required. Any volume of gas obtained in an assay can be corrected to standard temperature and pressure by multiplying by 100 and dividing by the number of c.c. in the corrector at the time the assay is made.
[5] It is best to use this form with a glass stopcock, or with an india-rubber tube and clip, after the manner of a Mohr's burette.
Acetic Acid, H[=A=c] or C2H4O2. (sp. gr. 1.044, containing 33 per cent. real acid).—An organic acid, forming a class of salts, acetates, which are for the most part soluble in water, and which, on ignition, leave the oxide or carbonate of the metal. It is almost always used in those cases where mineral acids are objectionable. To convert, for example, a solution of a substance in hydrochloric acid into a solution of the same in acetic acid, alkali should be added in excess and then acetic acid. Many compounds are insoluble in acetic acid, which are soluble in mineral acids, such as ferric phosphate, ferric arsenate, zinc sulphide, calcium oxalate, &c., so that the use of acetic acid is valuable in some separations. The commercial acid is strong enough for most purposes, and is used without dilution.
"Aqua Regia" is a mixture of 1 part by measure of nitric acid and 3 parts of hydrochloric acid. The acids react forming what is practically a solution of chlorine.[6] The mixture is best made when wanted, and is chiefly used for the solution of gold and platinum and for "opening up" sulphides. When solutions in aqua regia are evaporated, chlorides are left.
Bromine, Br. (sp. gr. 3.0). Practically pure bromine.—It is a heavy reddish-brown liquid and very volatile. It boils at 60° C., and, consequently, must be kept in a cool place. It gives off brown irritating vapours, which render its use very objectionable. Generally it answers the same purpose as aqua regia, and is employed where the addition of nitric acid to a solution has to be specially avoided. It is also used for dissolving metals only from ores which contain metallic oxides not desired in the solution.
"Bromine Water" is simply bromine shaken up with water till no more is dissolved.
Carbonic Acid, CO2.—A heavy gas, somewhat soluble in water; it is mainly used for providing an atmosphere in which substances may be dissolved, titrated, &c., without fear of oxidation. It is also used in titrating arsenic assays with "iodine" when a feeble acid[Pg 55] is required to prevent the absorption of iodine by the alkaline carbonate. It is prepared when wanted in solution, by adding a gram or so of bicarbonate of soda and then as much acid as will decompose the bicarbonate mentioned. When a quantity of the gas is wanted, it is prepared, in an apparatus like that used for sulphuretted hydrogen, by acting on fragments of marble or limestone with dilute hydrochloric acid.
Citric Acid (H3[=C=i] or C6H8O7.H2O) is an organic acid which occurs in colourless crystals, soluble in less than their weight of water. The solution must be freshly prepared, as it gets mouldy when kept. It forms a comparatively unimportant class of salts (citrates). It is used in the determination of phosphoric acid, chiefly for the purpose of preventing the precipitation of phosphates of iron and alumina by ammonia, and in a few similar cases. The commercial crystals are used; they should be free from sulphuric acid and leave no ash on ignition.
Hydrochloric Acid, HCl in water, (sp. gr. 1.16. It contains 32 per cent. of hydrogen chloride).—It is sometimes called "muriatic acid," and when impure, "spirit of salt." The acid solution should be colourless and free from arsenic, iron, and sulphuric acid. It forms an important family of salts, the chlorides. It is the best acid for dissolving metallic oxides and carbonates, and is always used by the assayer when oxidising agents are to be avoided. The acid is used without dilution when no directions are expressly given to dilute it. It has no action on the following metals: gold, platinum, arsenic, and mercury; it very slightly attacks antimony, bismuth, lead, silver, and copper. Tin is more soluble in it, but with difficulty; whilst iron, zinc, nickel, cobalt, cadmium, and aluminium easily dissolve with evolution of hydrogen and the formation of the lower chloride if the metal forms more than one class of salts. All the metallic oxides, except a few of the native and rarer oxides, are dissolved by it with the formation of chlorides of the metal and water.
Dilute Hydrochloric Acid is made by diluting the strong acid with an equal volume of water. This is used for dissolving precipitates obtained in the general course of analysis and the more easily soluble metals.
Hydrofluoric Acid, HF.—A solution in water may be purchased in gutta-percha or lead bottles. It is of variable strength and doubtful purity. It must always be examined quantitatively for the residue left on evaporation. It is used occasionally for the examination of silicates. It attacks silica, forming fluoride of silicon, which is a gas. When the introduction of another base will not interfere with the assay, the substance may be mixed in the platinum dish with fluoride of ammonium,[Pg 56] or of potassium, or of calcium, and hydrochloric acid, instead of treating it with the commercial acid. It is only required in special work. The fumes and acid are dangerous, and, of course, glass or porcelain vessels cannot be used with it.
Iodine, I.—This can be obtained in commerce quite pure, and is often used for standardising. It is very slightly soluble in water, but readily dissolves in potassium iodide solution. It closely resembles chlorine and bromine in its properties, and can be used for dissolving metals without, at the same time, attacking any oxide which may be present. It is chiefly used as an oxidizing agent in volumetric work, being sharp in its reactions and easily detected in minute quantities. It cannot be used in alkaline solutions, since it reacts with the hydrates, and even with the carbonates, to form iodides and iodates. Iodine is soluble in alcohol.
Nitric Acid, HNO3. (Sp. gr. 1.42; boiling point 121° C.; contains 70 per cent. by weight of hydrogen nitrate).—It is convenient to remember that one c.c. of this contains 1 gram of real acid. It combines the properties of an acid and of an oxidising agent. One c.c. contains 0.76 gram of oxygen, most of which is very loosely held, and easily given up to metals and other oxidisable substances. Consequently it will dissolve many metals, &c., upon which hydrochloric acid has no action. All sulphides (that of mercury excepted) are attacked by it, and for the most part rendered soluble. It has no action on gold or platinum, and very little on aluminium. The strong acid at the ordinary temperature does not act on iron or tin; and in most cases it acts better when diluted. Some nitrates being insoluble in nitric acid, form a protecting coat to the metal which hinders further action. Where the strong acid does act the action is very violent, so that generally it is better to use the dilute acid. When iron has been immersed in strong nitric acid it not only remains unacted on, but assumes a passive state; so that if, after being wiped, it is then placed in the dilute acid, it will not dissolve. Tin and antimony are converted into insoluble oxides, while the other metals (with the exception of those already mentioned) dissolve as nitrates. During the solution of the metal red fumes are given off, which mainly consist of nitrogen peroxide. The solution is often coloured brown or green because of dissolved oxides of nitrogen, which must be got rid of by boiling. Generally some ammonium nitrate is formed, especially in the cases of zinc, iron, and tin, when these are acted on by cold dilute acid. Sulphur, phosphorus, and arsenic are converted into sulphuric, phosphoric, and arsenic acids respectively, when boiled with the strong acid.[Pg 57]
Dilute Nitric Acid.—Dilute 1 volume of the strong acid with 2 of water.
Oxalic Acid, H2O or (H2C2O4.2H2O.)—This is an organic acid in colourless crystals. It forms a family of salts—the oxalates. It is used in standardising; being a crystallised and permanent acid, it can be readily weighed. It is also used in separations, many of the oxalates being insoluble. For general use make a 10 per cent. solution. Use the commercially pure acid. On ignition the acid should leave no residue.
Sulphuretted Hydrogen. Hydrosulphuric acid, SH2.—A gas largely used in assaying, since by its action it allows of the metals being conveniently classed into groups. It is soluble in water, this liquid dissolving at the ordinary temperature about three times its volume of the gas. The solution is only useful for testing. In separations, a current of the gas must always be used. It is best prepared in an apparatus like that shown in fig. 32, by acting on ferrous sulphide with dilute hydrochloric acid. When iron has to be subsequently determined in the assay solution, the gas should be washed by bubbling it through water in the smaller bottle; but for most purposes washing can be dispensed with. The gas is very objectionable, and operations with it must be carried out in a cupboard with a good draught. When the precipitation has been completed, the apparatus should always be washed out. The effect of this acid on solutions of the metals is to form sulphides. All the metallic sulphides are insoluble in water; but some are soluble in alkaline, and some in acid, solutions. If sulphuretted hydrogen is passed through an acid solution containing the metals till no further precipitation takes place, a precipitate will be formed containing sulphides insoluble in the acid. On filtering, adding ammonia (to render the filtrate alkaline), and again passing the gas, a further precipitate will be obtained, consisting of sulphides insoluble in an alkaline solution, but not precipitable in an acid one; the filtrate may also contain sulphides not precipitable in an acid solution, which are soluble in an alkaline one; these will be thrown down on neutralising. Again, the metals precipitated in the acid solution form sulphides which may be divided into groups, the one consisting of those which are soluble, and the other of those which are not soluble, in alkalies. This classification is shown in the following summary:[Pg 58]—
1. Precipitable in an acid solution.
(a) Soluble in Alkalies.—Sulphides of As, Sb, Sn, Au, Pt, Ir, Mo, Te, and Se.
(b) Insoluble in Alkalies.—Sulphides of Ag, Pb, Hg, Bi, Cu, Cd, Pd, Rh, Os, and Ru.
2. Not precipitated in an acid solution, but thrown down in an alkaline one.
Sulphides of Mn, Zn, Fe, Ni, Co, In, Tl, and Ga.
These can again be divided into those which are dissolved by dilute acids and those which are not.
3. Not precipitated in an acid or alkaline solution, but thrown
down on neutralising the latter.
Sulphides of V and W.
Sulphuretted hydrogen is a strong reducing agent. Ferric salts are thereby quickly reduced to ferrous; in hot solutions nitric acid is decomposed. These changes are marked by a precipitation of sulphur, and the student must be careful to pass the gas sufficiently long, and not be too hasty in concluding that no sulphide will form because it does not at once make its appearance. The best indication that it has been passed long enough is the smell of the gas in the solution after shaking.
Sulphurous Acid, H2SO3.—The reagent used may be regarded as a saturated solution of sulphur dioxide in water. It may be purchased, and keeps for a long time. It may be made by heating copper with sulphuric acid and passing the gas formed into water. The heat should be withdrawn when the gas is coming off freely. It is used as a reducing agent, and should not be diluted.
Sulphuric Acid, H2SO4. (Sp. gr. 1.84, containing 96 per cent. of real acid, H2SO4.)—This acid forms insoluble sulphates with salts of lead, strontium, and barium. It has a high boiling point, 290° C., and, when evaporated with salts of the more volatile acids, converts them into sulphates. When nitrates or chlorides are objectionable in a solution, evaporation with sulphuric acid removes them. In working with this acid caution is necessary, since, on mixing with water, great heat is evolved; and, if either the acid or water has been previously heated, a serious accident may result. In diluting the acid it should be poured into cold water. Glass vessels containing boiling sulphuric acid should be handled as little as possible, and should not be cooled under the tap. The action of diluted sulphuric acid on metals closely resembles that of dilute hydrochloric acid. Magnesium, aluminium, iron, zinc, nickel, cobalt, manganese, and cadmium dissolve, with evolution of hydrogen, in the cold acid, or when warmed. The action of hot and strong sulphuric acid is[Pg 59] altogether different; it acts as an oxidising agent, and is itself reduced to sulphur dioxide or even to sulphur. The following metals are attacked in this way:—copper, bismuth, mercury, silver, antimony, tin, and lead. Gold, platinum, and arsenic are not affected. This property is made use of in parting silver from gold and platinum. Metallic sulphides are similarly attacked; but this method of opening up minerals has the disadvantage of giving rise to the formation of anhydrous sulphates of iron, &c., which are not readily dissolved when afterwards diluted. The use of sulphuric acid in assaying is (for these reasons) to be avoided. Its chief use is as a drying agent, since it has a strong affinity for water. Air under a bell jar may be kept dry by means of a basin of sulphuric acid, and gases bubbled through it are freed from water-vapour.
Dilute Sulphuric Acid.—This is made by diluting 1 volume of the strong acid with 4 of water.
Tartaric Acid, H2[=T] or C4H6O6.—A crystallised organic acid, soluble in less than its own weight of water, or in less than three parts of alcohol. It is used for the same purposes as citric acid is. The solution is made when required.
Alcohol, C2H6O. (Commercial alcohol of sp. gr. 0.838; it contains 84 per cent. by weight of alcohol.)—It should burn with a non-luminous flame and leave no residue. It is used for washing precipitates where water is inapplicable, and for facilitating drying.
Ammonia, NH3. (Commercial ammonia, a solution having a sp. gr. of 0.88 to 0.89, and containing about 33 per cent. of ammonia.)—It is used as an alkali (more commonly than soda or potash), since an excess of it is easily removed by boiling. The salts of ammonium formed by it may be removed by igniting, or by evaporating in a porcelain dish with an excess of nitric acid. It differs in a marked way from soda or potash in its solvent action on the oxides or hydrates of the metals. Salts of the following metals are soluble in an ammoniacal solution in the presence of ammonic chloride:—copper, cadmium, silver, nickel, cobalt, manganese, zinc, magnesium, sodium, potassium, and the alkaline earths.
Dilute Ammonia is made by diluting 1 vol. of commercial ammonia with 2 of water. The dilute ammonia is always used; but in assays for copper a stronger solution (1 of strong ammonia to 1 of water) is required.
Ammonic Carbonate (Am2CO3) is prepared by dissolving one part of the commercial sesquicarbonate of ammonia in four parts of water, and adding one part of strong ammonia.[Pg 60]
Ammonic Bicarbonate (HAmCO3) is prepared by saturating a solution of the sesquicarbonate of ammonia with carbon dioxide.
Ammonic Chloride, AmCl.—Use the commercial salt in a 20 per cent. solution in water. The salt should leave no residue on ignition.
Ammonic Molybdate.—The solution is prepared as follows:—Dissolve 100 grams of the powdered commercial salt in 200 c.c. of dilute ammonia, and pour the solution in a slow stream into 750 c.c. of dilute nitric acid; make up to 1 litre, and allow the mixture to settle before using. It is used for the purpose of separating phosphoric oxide from bases and from other acids, and also as a test for phosphates and arsenates. In using this solution the substance must be dissolved in nitric acid, and a considerable excess of the reagent added (50 c.c. is sufficient to precipitate 0.1 gram P2O5); when the phosphate is in excess no precipitate will be got. The precipitate is phospho-molybdate of ammonia.
Ammonic Nitrate (AmNO3) is used in the separation of phosphoric oxide by the molybdate method, and occasionally for destroying organic matter. It is soluble in less than its own weight of water. The solution is made when wanted.
Ammonic Oxalate (Am2C2O4.2H2O) is used chiefly for the separation of lime. The solution is made by dissolving 15 grams of the salt in 100 c.c. of water.
Ammonic Sulphide may be purchased in the state of a strong solution. It is yellow, and contains the disulphide, S2Am2. It serves the same purpose as is obtained by passing a current of sulphuretted hydrogen through an ammoniacal solution; but has the disadvantage of loading the solution with sulphur, which is precipitated when the solution is subsequently acidified. It is useful for dissolving the lower sulphide of tin (SnS).
Baric Carbonate (BaCO3) is sometimes used for precipitating the weaker bases. It should be prepared when wanted by precipitating a solution of baric chloride with ammonic carbonate and washing. The moist precipitate is used without drying.
Baric Chloride, BaCl2.2H2O.—A crystallised salt, soluble in 2-1/2 parts of water. It is used for the detection and separation of sulphates. Make a 10 per cent. solution.
"Black Flux."—A mixture of finely divided carbon with carbonate of potash or with carbonates of potash and soda. It is prepared by heating tartar or "rochelle salt" until no more combustible gas is given off. One gram will reduce about 2 grams of lead from litharge.
Borax, Na2B4O7.10H2O.—It is chiefly used as a flux in dry assaying, as already described. It is also used in testing before[Pg 61] the blowpipe; many metallic oxides impart a characteristic colour to a bead of borax in which they have been fused.
Calcium Chloride.—The crystallised salt is CaCl2.6H2O; dried at 200° C. it becomes CaCl2.2H2O, and when fused it becomes dehydrated. The fused salt, broken into small lumps, is used for drying gases. It combines with water, giving off much heat; and dissolves in a little more than its own weight of water. Strong solutions may be used in baths in which temperatures above the boiling-point of water are required. One part of the salt and 2 of water give a solution boiling at 112°, and a solution of 2 parts of the salt in 1 of water boils at 158°. The salt is very little used as a reagent.
Calcium Fluoride or "Fluor Spar," CaF2.—The mineral is used as a flux in dry assaying; it renders slags which are thick from the presence of phosphates, &c., very fluid. Mixed with hydrochloric acid it may sometimes be used instead of hydrofluoric acid.
Calcium Carbonate, CaCO3.—It is precipitated in a pure state by ammonic carbonate from a solution of calcium chloride. It is used for standardising. In the impure state, as marble or limestone, it is used in the preparation of carbonic acid.
Calcium Hydrate or "Lime Water."—This is used in testing for carbon dioxide and in estimating the amount of that gas present in air. It may be made by slaking quicklime and digesting the slaked lime with water. One hundred c.c. of water at 15° C. dissolves 0.1368 grams of the hydrate (CaH2O2), and hot water dissolves still less. "Milk of lime" is slaked lime suspended in water.
Cobalt Nitrate (Co(NO3)2.6H2O) is used in a 10 per cent. solution for the detection of oxides of zinc, aluminium, &c.; on ignition with which it forms characteristically coloured compounds.
Copper, Cu.—Pure copper, as obtained by electrolysis, can be purchased. This only should be used.
Copper Oxide, CuO.—It occurs as a black, heavy, and gritty power, and is used for the oxidation of carbon and hydrogen in organic substances. It should be ignited and cooled out of contact with air just before using, since it is hygroscopic. Oxide of copper which has been used may be again utilised after calcination.
Copper Sulphate (CuSO4.5H2O) contains 25.4 per cent. of copper. It is used in the outer cell of a Daniell-battery. The commercial salt is used for this purpose. The re-crystallised and pure salt is used for preparing the anhydrous sulphate, which is used for detecting moisture in gases. For this purpose it is[Pg 62] dried at 200° C. till no trace of green or blue colour remains. It must be prepared when wanted. It may be conveniently used in the form of pumice-stone, saturated with a solution of the salt and dried. Traces of moisture develop a green colour.
Ferric Chloride, Fe2Cl6. (When crystallised, Fe2Cl6.6H2O.)—The solution is prepared as described under iron. The commercial salt contains arsenic, and, since the chief use of ferric chloride is for the determination of this substance, it must be purified (see under Arsenic).
Ferric Sulphate (Fe2(SO4)3) is a yellowish white deliquescent salt. It is used as an indicator in volumetric silver assaying, and for the separation of iodine from bromine. It may be purchased as iron alum, Am2Fe2(SO4)4.24H2O. But it is best prepared by adding strong sulphuric acid to ferric hydrate in equivalent proportions. Use it as a solution containing 2 or 3 per cent. of iron.
Ferrous Sulphate, FeSO4.7H2O.—The granulated form is best, and can be purchased pure. It is used for standardising. It keeps better in crystals than in solution. It is readily soluble in water, but the solution is best made with the help of a little free acid. As a re-agent use a 10 per cent. solution. The crystals should be clear bluish-green; if their colour is dark green, brown, or blue, they should be rejected.
Ferrous Sulphide (FeS) is used for the preparation of sulphuretted hydrogen. It may be purchased and broken in small lumps, nut-size, for use.
"Fusion Mixture" (K2CO3.Na2CO3) is a mixture of potassic and sodic carbonates in the proportions of 13 of the former to 10 of the latter, by weight. It is hygroscopic. A mixture of the bicarbonates is better, being purer and less apt to get damp.
Gallic Acid (C7H6O5.H2O) is an organic acid, occurring as a pale fawn-coloured crystalline powder, soluble in 100 parts of cold water, or in 3 parts of boiling water. It is used for the determination of antimony. A 10 per cent. solution in warm water is made when required.
Hydrogen (H) is a gas. It is obtained by acting on zinc with dilute hydrochloric or sulphuric acid. It is used as a reducing agent, and for providing an atmosphere free from oxygen. It reduces metallic oxides at a high temperature. It must be freed from water; and special precautions should be taken to prevent an admixture with air. It is generally required in a current which can be continued for an hour or more without interruption. The preparation can be conveniently carried out in the apparatus shown (fig. 33). A quart bottle is half filled with sheet zinc, and connected with bulbs filled with sulphuric acid, and with[Pg 63] a calcium chloride tube. The last is connected with the apparatus through which the gas has to be passed. Dilute hydrochloric acid mixed with a few cubic centimetres (20 c.c. to 1 pint) of stannous chloride sol. to fix any dissolved oxygen, is placed in the funnel, and let into the bottle by opening the stopcock when required. Care must be taken to let the hydrogen escape for some time before starting the reduction.
Gold, Au.—Gold, obtained by cupelling and "parting," is for most purposes sufficiently pure. It is best kept in the shape of foil. When the purer metal is required, gold should be dissolved in aqua regia, the solution evaporated to a paste, diluted, allowed to stand, and filtered. The filtered solution is acidified with hydrochloric acid, warmed, and precipitated with sodium sulphite. The precipitate is collected, washed, and fused on charcoal.
Iron, Fe.—The soft wire (thin) is used for standardising. Rods are used in dry assays as a desulphurising agent. Steel must not be used, since it is not pure, and contains a variable amount of iron.
Lead, Pb.—Granulated lead or lead-foil is used in the dry assay for silver and gold, and in the preparation of lead salts. It can be obtained very pure, but always contains more or less silver, 1 or 2 milligrams in 100 grams. The amount of silver it contains must be determined and recorded.
Lead Acetate (Pb[=A=c]2.3H2O, or Pb(C2H3O2)2.3H2O) is used as a test, specially for the detection and estimation of sulphuretted hydrogen. Prepare a 10 per cent. solution for use.
Lead Nitrate (Pb(NO3)2) can be purchased pure. It is used for standardising.
Lead Dioxide (PbO2) occurs as a dark-brown powder. It is used as an oxidizing agent and for absorbing sulphurous oxide. It can be prepared by digesting red lead with warm dilute nitric acid; washing and drying the residue.
"Litharge," PbO.—It can be purchased as a yellow heavy powder. It is used in dry assaying as a flux, as a desulphurising agent, and also as a source of lead. It always contains some silver, the amount of which must be determined.
Litmus.—This is an organic colouring matter which is turned red by acids and blue by alkalies. For ordinary purposes it is best used as litmus paper, which may be purchased in small books. A solution is prepared by digesting 15 or 20 grams of the commercial litmus in 100 c.c. of water on the water bath. After being allowed to settle, it is filtered and made just faintly red with[Pg 64] acetic acid. Then there is added a drop or two of a solution of soda and 10 c.c. of alcohol. It should be kept in a loosely-covered bottle.
Magnesia, MgO.—It may be purchased as "calcined magnesia." It is used for making "magnesia mixture," and should be kept in a corked wide-mouthed bottle.
"Magnesia Mixture."—Dissolve 22 grams of magnesia in about a quarter of a litre of dilute hydrochloric acid, avoiding excess. Add 5 grams of magnesia, boil, and filter. Add 300 grams of ammonic chloride, and 250 c.c. of strong ammonia; and dilute with water to 2 litres. It should be kept in a stoppered winchester.
Magnesium Sulphate, MgSO4.7H2O.—It can be purchased very pure, and is occasionally used as a standard salt.
Manganese Dioxide, MnO2.—It is used in the preparation of chlorine. The commercial article is not pure, but is sufficiently so for this purpose.
Marble, CaCO3.—Fragments of the white crystalline variety only should be used. It is used as a source of lime and of carbon dioxide.
Mercury, Hg.—This can be purchased pure. It should have a bright surface, flow without a tail, and leave no residue on ignition. It is used as a standard; for amalgamation; and as a confining liquid in gas analysis.
Mercuric Chloride (HgCl2) may be purchased pure. Make a 5 per cent. solution in water. It is used for destroying an excess of stannous chloride; for removing sulphuretted hydrogen from solution; and as a test for stannous salts.
Microcosmic Salt, HAmNaPO4.8H2O.—When fused NaPO3 is formed. It is used in testing for metallic oxides and silica before the blowpipe. The crystals are sometimes used as a standard for phosphoric acid.
"Nessler's Solution."—Mode of preparation: Dissolve 35 grams of potassium iodide in 100 c.c. of water; dissolve 17 grams of mercuric chloride in 300 c.c. of water, and pour this solution into that of the iodide till a permanent precipitate is produced; make up to 1 litre with a 20 per cent. solution of potash; add mercuric chloride till a precipitate is again formed; allow to settle and decant. It is used for detecting ammonia.
Nitre.—This is potassium nitrate.
Platinum Chloride, 2HCl.PtCl4. (In the crystallised form it has 6H2O).—It may be made as follows:—Take 5 grams of clean platinum scrap and dissolve in a flask at a gentle heat in 50 c.c. of hydrochloric acid with the occasional addition of some nitric acid; evaporate to a paste; and then dissolve in 100 c.c. of water. It is used for separating and determining potassium.[Pg 65]
Phenolphthalein is an organic compound used as an indicator; more especially in determining the weaker acids, it cannot be used in the presence of ammonia. Dissolve half a gram in 100 c.c. of dilute alcohol.
Potassium Bicarbonate, KHCO3.—It may be purchased pure; on ignition it leaves the carbonate, K2CO3, which may be used as a standard.
Potassium Cyanide, KCN.—It is used in the dry assay as a reducing agent. The commercial salt is very impure. Purchase that sold as potassic cyanide (gold) which contains about 95 per cent. of KCN. It is used for copper assaying and occasionally in separation. Make a 10 per cent. solution when wanted.
Potassium Bichromate, K2Cr2O7. It may be purchased nearly pure. It is used as an oxidising agent, for determining iron; and as a test solution. For this last purpose a 10 per cent. solution is prepared.
Potassium Chlorate (KClO3) can be purchased pure. It is used with hydrochloric acid as a substitute for aqua regia.
Potassium Ferrocyanide (K4Fe(CN)6.3H2O), or "yellow prussiate of potash," is used as a test; as an indicator; and for the determination of zinc. Make a 5 per cent. solution.
Potassium Ferricyanide (K6Fe2(CN)12), or "red prussiate of potash," is used for testing; and as an indicator. Make a 5 per cent. solution when wanted, as it decomposes on keeping.
Potassium Hydrate, KHO. Purchase that purified with alcohol. It is an alkali, and is used for absorbing carbonic acid, &c.
Potassium Iodide, KI. It may be purchased nearly pure. It is used as a test and for dissolving iodine. It should be used in a 10 per cent. solution freshly made. The solution decomposes on exposure to light, with separation of iodine.
Potassium Nitrate (KNO3) can be purchased pure. It is used in the dry way as an oxidizing agent. It is very fusible. It decomposes at a low temperature into potassium nitrite (KNO2) and free oxygen; and at a higher temperature leaves potash (K2O). It oxidizes sulphur and carbon with explosive violence. This action may be moderated by mixing the nitre with carbonate of soda, common salt, or some other inert body.
Potassium Nitrite, KNO2.—The commercial article is not pure, but is sufficiently so for the purpose required. A saturated solution is used in the separation of cobalt; the solution is made when wanted.
Potassium Permanganate, KMnO4.—This salt can be purchased sufficiently pure. It is much used as an oxidizing agent.
Potassium Bisulphate (KHSO4) is used as a dry reagent for opening up minerals. It fuses; and at a much higher temperature[Pg 66] is converted into potassium sulphate with loss of sulphuric acid.
Potassium Sulphocyanate (KCNS) is used for the detection and determination of traces of ferric iron; as also in the separation of silver and copper from some of the other metals. Make a 10 per cent. solution. It should show no colour on the addition of hydrochloric acid.
"Red Lead" (Pb3O4) is used in the dry assay as a flux instead of litharge, from which it differs in containing a little more oxygen. When acted on by nitric acid a brown residue of lead dioxide is left, nitrate of lead going into solution. Like litharge it always carries silver; about 2 milligrams in 100 grams.
Silver, Ag.—Pure silver in foil is required as a standard. It may be prepared as follows:—Dissolve scrap silver in dilute nitric acid and decant off from any residue; dilute the solution with hot water and add hydrochloric acid until there is no further precipitate, stir; allow the precipitate to settle; decant and wash; dry the precipitate, mix it with twice its bulk of carbonate of soda and fuse the mixture in a crucible until tranquil; clean the button and roll or hammer it into foil.
Sodium Acetate, NaC2H3O2.3H2O.—The crystals may be purchased sufficiently pure. Make a 20 per cent. solution in water. It is used for replacing mineral acids by acetic acid.[7]
Sodium Acetate and Acetic Acid.—A solution is used in the determination of phosphates and arsenates; 100 grams of the salt is dissolved in 500 c.c. of acetic acid, and diluted with water to one litre.
Sodium Bicarbonate (NaHCO3)is used as a flux in dry methods. On ignition it leaves the carbonate (Na2CO3), which is used as a standard reagent. Make a 20 per cent. solution of the carbonate for use. It should be free from chlorides or sulphates, or if impure the amount of impurities must be determined.
Sodium Hydrate, NaHO. It may be purchased in sticks, which should be kept in a well-corked bottle. It is sometimes called "caustic soda." It is a strong alkali. It is used for neutralizing acid solutions and for separations where ammonia is unsuitable. Make a 5 per cent. solution for use.
Sodium Hyposulphite, Na2S2O8.5H2O.—It may be purchased pure. It is generally known as "hypo." It is used as a standard.
Sodium Sulphite (Na2SO3.7H2O) is used as a reducing agent.
Sodium Phosphate, Na2HPO4.12H2O. The crystals may be purchased pure, but they effloresce in dry air with loss of water.[Pg 67] It is used as a standard and for precipitating magnesia, &c. Make a 10 per cent. solution.
Stannous Chloride, SnCl2.2H2O.—The crystals are best purchased. If kept dry and free from air they are fairly permanent. A solution is made by dissolving 20 grams in 10 c.c. of hydrochloric acid and diluting to 1 litre. The solution is not permanent. It is a strong reducing agent, and is chiefly used in solution for this purpose.
Tin, Sn.—Grain tin should be purchased. It is not pure, but contains 99.5 per cent. of the metal. The chief impurity is copper. It can be used as a standard. When acted on with hot hydrochloric acid it slowly dissolves (more rapidly in contact with platinum) and forms stannous chloride.
Uranium Acetate, UO2(C2H3O2)2.H2O.—It is best purchased in crystals. The solution is used for the determination of phosphates and arsenates. A solution of 3 per cent. strength is occasionally used as an indicator.
Uranium Nitrate, UO2(NO3)2.6H2O.—This salt is very soluble in water and is sometimes used instead of the acetate, which is somewhat difficult to dissolve.
"Water," H2O.—Spring or well water is sufficiently pure for most purposes, 100 c.c. will leave a residue of from 10 to 30 milligrams, so that where a salt has to be dissolved out, evaporated, and weighed it should be replaced by distilled water. Rain water, melted snow, &c., always leave less residue than spring water; but in other respects they are often dirtier. Distilled water is best prepared in the office, a glass or tin condenser being used.
Zinc, Zn.—It is sold in a granulated form or in sticks. It generally contains over 1 per cent. of lead, with a little iron and arsenic. It is used for separating metals from their solutions, and generally as a reducing agent. For the preparation of hydrogen, and in most other cases, scrap sheet zinc may be used.
Zinc Oxide, ZnO.—The commercial oxide sometimes contains carbonate.
Zinc Sulphate, ZnSO4.7H2O.—It is occasionally used as a standard, and can be purchased nearly pure.
Formulæ and equations are a kind of short hand for expressing briefly and in the language of the atomic theory the facts of chemical composition and reaction. The convenience of this method of expressing the facts justifies a short description of it here.
On comparing the percentage composition of a series of compounds the proportions in which the elements combine appears to be regulated by no simple law. For example:
| Realgar. | Orpiment. | Mispickel. | Pyrites. | |
| Arsenic | 71.4 | 60.9 | 46.0 | — |
| Sulphur | 28.6 | 39.1 | 19.6 | 53.3 |
| Iron | — | — | 34.4 | 46.7 |
| ——— | ——— | ——— | ——— | |
| 100.0 | 100.0 | 100.0 | 100.0 |
But if in these examples the composition is calculated, not on 100 parts, but on 107, 246, 163, and 120 parts respectively, evidence of a simple law becomes apparent.
| Realgar. | Orpiment. | Mispickel. | Pyrites. | |
| Arsenic | 75.0 | 150.0 | 75.0 | — |
| Sulphur | 32.0 | 96.0 | 32.0 | 64.0 |
| Iron | — | — | 56.0 | 56.0 |
| ——— | ——— | ——— | ——— | |
| 107.0 | 246.0 | 163.0 | 120.0 |
It will be seen that the proportion of arsenic is 75 or twice 75, that of iron is 56, and that of sulphur 32 or some simple multiple of 32. The series of examples might be extended indefinitely, and it would still be found that the "combining proportions" held good. The number 75 is spoken of as the "combining weight," or, more frequently, as the "atomic weight" of arsenic. Similarly 56 is the atomic weight of iron, and 32 the atomic weight of sulphur. The importance of this law of chemical combination is altogether independent of the atomic theory; but this theory furnishes the simplest explanation of the facts. According to it a chemical compound is made up of exactly similar groups of particles. The[Pg 69] particles of each elementary substance are all alike, but differ from those of other elements in weight. Ultimate particles are called atoms, and the groups of atoms are called molecules. The atomic weight of any particular element is the weight of its atom compared with the weight of an atom of hydrogen. The atom of sulphur, for instance, is 32 times as heavy as the atom of hydrogen, and the atomic weight of sulphur is 32. The molecular weight is the sum of the atomic weights of the group. The molecule of pyrites contains two atoms of sulphur and one of iron: on referring to the table of atomic weights it will be seen that the atomic weights are—sulphur 32, and iron 56. The molecular weight, therefore, is 32 + 32 + 56—that is, 120. The meaning of this is, 120 parts by weight of iron pyrites contain 64 parts of sulphur and 56 parts of iron; and this is true whether the "parts by weight" be grains or tons.
The symbol or formula of an atom is generally the initial letter or letters of the Latin or English name of the substance. The atom of hydrogen is written H, that of oxygen O, of sulphur S, of iron (ferrum) Fe, and so on. A list of these symbols is given in the table of atomic weights.
The formula of a molecule is obtained by placing together the symbols of the contained atoms. Thus, Fe represents an atom of iron, S an atom of sulphur, while FeS represents the molecule of sulphide of iron as containing one atom of each element.
When more than one atom of an element is present this is shown by writing a figure under and after the symbol; thus, FeS2 represents a molecule with one atom of iron and two atoms of sulphur, Fe2S3 similarly shows one with two atoms of iron and three of sulphur. When a group of atoms is enclosed in brackets, a figure after and under the bracket multiplies all within it; for example, Pb(NO3)2 is another way of writing PbN2O6. Sometimes it is convenient to represent the atoms of a molecule as divided into two or more groups; this may be done by writing the formulæ of the groups, and separating each simple formula by a full stop. Slaked lime, for instance, has the formula CaH2O2; or, as already explained, we may write it Ca(HO)2; or, if for purposes of explanation we wished to look on it as lime (CaO) and water (H2O), we could write it CaO.H2O. A plus sign (+) has a different meaning; CaO + H2O indicates quantities of two substances, water and lime, which are separate from each other. The sign of equality (=) is generally used to separate a statement of the reagents used from another statement of the products of the reaction; it may be translated into the word "yields" or "becomes." The two statements form an equation.
Ignoring the quantitative relation, the meaning of the equation[Pg 70] CaO + H2O = CaO.H2O is: "lime and water yield slaked lime." By referring to a table of atomic weights we can elicit the quantitative relations thus:—
| CaO | + | H2O | = | CaH2O2 | |
| ↓ | ↓ | ↓ | |||
| Ca = 40 | H2 = 2 | = 1×2 | Ca = 40 | ||
| O = 16 | O = 16 | H2 = 2 | = 1×2 | ||
| —— | —— | O2 = 32 | = 16×2 | ||
| 56 | 18 | —— | |||
| 74 |
Or, putting it in words, 56 parts of lime combine with 18 parts of water to form 74 parts of slaked lime. This equation enables one to answer such a question as this:—How much lime must be used to produce 1 cwt. of slaked lime? for, if 74 lbs. of slaked lime require 56 lbs. of lime, 112 lbs. will require (56 × 112)/74, or about 84-3/4 lbs.
As another example having a closer bearing on assaying take the following question:—"In order to assay 5 grams of 'black tin' (SnO2) by the cyanide process, how much potassic cyanide (KCN) will be required?" The reaction is
| SnO2 | + | 2KCN | = Sn + 2KCNO |
| ↓ | ↓ | ||
| Sn = 118 | K = 39 | ||
| O2 = 32 | C = 12 | ||
| —— | N = 14 | ||
| 150 | —— | ||
| 65 | ×2 = 130 |
What is sought for here is the relation between the quantities of SnO2 and KCN. Note that a figure before a formula multiplies all that follows up to the next stop or plus or equality sign. The question is now resolved to this: if 150 grams of oxide of tin require 130 grams of cyanide, how much will 5 grams require?
150 : 130 :: 5 : x
x = 4.33 grams.
A problem of frequent occurrence is to find the percentage composition of a substance when its formula has been given. For example: "What percentage of iron is contained in a mineral having the formula 2Fe2O3.3H2O?" Bringing this formula together we have Fe4H6O9. Find the molecular weight.
| Fe4 | = 224 | = 56×4 |
| H6 | = 6 | = 1×6 |
| O9 | = 144 | = 16×9 |
| —— | ||
| 374 |
Then we get: 374 parts of the mineral contain 224 of iron. How much will 100 contain?
374 : 224 :: 100 : x
x = 59.89.
And the answer to the question is 59.89 per cent.
Again, suppose the question is of this kind:—"How much crystallised copper sulphate (CuSO4.5H2O) will be required to make 2 litres of a solution, 1 c.c. of which shall contain 0.0010 gram of copper?"
A litre is 1000 c.c., so, therefore, 2 litres of the solution must contain 0.001 gram × 2000, or 2 grams. How much crystallised copper sulphate will contain this amount of metal?
| Cu | = 63.3 | |
| S | = 32.0 | |
| O4 | = 64.0 | = 16×4 |
| 5H2O | = 90.0 | = 18×5 |
| ———— | ||
| 249.3 |
If 63.3 grams of copper are contained in 249.3 grams of sulphate, in how much is 2 grams contained.
63.3 : 249.3 :: 2 grams : x
x = 7.8769 grams.
The answer is, 7.8769 grams must be taken.
As a sample of another class of problem similar in nature to the last (but a little more complicated) take the following:—"What weight of permanganate of potash must be taken to make 2 litres of a solution, 100 c.c. of which shall be equivalent to 1 gram of iron?" In the first place the 2 litres must be equivalent to 20 grams of iron, for there are 20 × 100 c.c. in two litres. In the titration of iron by permanganate solution there are two reactions. First in dissolving the iron
Fe + H2SO4 = FeSO4 + H2
↓
56
and second, in the actual titration,
10FeSO4 + 2KMnO4 + 9H2SO4= 2MnSO4 + 5Fe2(SO4)3 + 2KHSO4 + 8H2O
↓
K = 39
Mn = 55
O4= 64
——
158 × 2 = 316
As before, attention is confined to the two substances under[Pg 72] consideration—viz., Fe and KMnO4. In the second equation, we find 316 parts of the permanganate are required for 10 molecules of FeSO4; and in the first equation 56 parts of iron are equivalent to one molecule of FeSO4, therefore 560 of iron are equivalent to 316 of permanganate; and the question is, How much of the permanganate will be equivalent to 20 grams of iron?
560 : 316 :: 20 grams : x.
x = 11.286 grams.
The answer is 11.286 grams.
Very similar to this last problem is the question suggested under the head "Indirect Titration" (p. 43). "If 100 c.c. of the standard permanganate solution are equivalent to 1 gram of iron, how much peroxide of manganese will they be equivalent to?" The equation for dissolving the iron is already given; the second equation is
2FeSO4 + MnO2 + 2H2SO4 = Fe2(SO4)2 + MnSO4 + 2H2O
↓
Mn = 55
O2 = 32
——
87
It will be seen that 87 grams of peroxide of manganese are equivalent to 112 grams of iron. How much then is equivalent to 1 gram of iron?
112 : 87 :: 1 gram : x
x = 0.7767 gram.
It is sometimes convenient to calculate the formula of a substance from its analysis. The method of calculating is shown by the following example. Required the formula of a mineral which gave the following figures on analysis:—
| Cupric oxide (CuO) | 10.58 |
| Ferrous oxide (FeO) | 15.69 |
| Zinc oxide (ZnO) | 0.35 |
| Sulphuric oxide (SO2) | 28.82 |
| Water (H2O) | 44.71 |
| —————— | |
| 100.15 |
First find the molecular weights of CuO, FeO, &c., and divide the corresponding percentages by these figures. Thus, CuO = 63.3+16 = 79.3 and 10.58 divided by 79.3 gives 0.1334. Similarly FeO = 56+16 = 72 and 15.69 divided by 72 gives 0.2179. Treated in the same way the oxide of zinc, sulphuric oxide and water give as results 0.0043, 0.3602 and 2.484.
Classify the results as follows:[Pg 73]—
| Bases. | Acids. | Water. |
| CuO 0.1334 | SO3 0.3602 | H2O 2.484 |
| FeO 0.2179 | ||
| ZnO 0.0043 | ||
| —————————— | —————————— | —————————— |
| RO 0.3556 | RO3 0.3602 | R2O 2.484 |
The figures 0.3556, 0.3602 and 2.484 should be then divided by the lowest of them—i.e., 0.3556; or where, as in this case, two of the figures are very near each other the mean of these may be taken—i.e., 0.3579. Whichever is taken the figures got will be approximately 1, 1 and 7. The formula is then RO.SO3.7H2O in which R is nearly 2/5ths copper, 3/5ths iron and a little zinc.
This formula requires the following percentage composition, which for the sake of comparison is placed side by side with the actual results.
| Calculated. | Found. | |
| Cupric oxide | 11.29 | 10.58 |
| Ferrous oxide | 15.37 | 15.69 |
| Zinc oxide | nil | 0.35 |
| Sulphuric oxide | 28.47 | 28.82 |
| Water | 44.84 | 44.71 |
| ——— | ——— | |
| 99.97 | 100.15 |
Trimming the results of an analysis to make them fit in more closely with the calculations from the formula would be foolish as well as dishonest. There can be no doubt that the actual analytical results represent the composition of the specimen much more closely than the formula does; although perhaps other specimens of the same mineral would yield results which would group themselves better around the calculated results than around those of the first specimen analysed. It must be remembered that substances are rarely found pure either in nature or in the arts; so that in most cases the formula only gives an approximation to the truth. In the case of hydrated salts there is generally a difficulty in getting the salt with exactly the right proportion of water.
The following calculations may be made:—
1. Calculate standards in the following cases—
(a) Silver taken, 1.003 gram. Standard salt used, 100.15 c.c.
(b) Iron taken, 0.7 gram. Bichromate used, 69.6 c.c.
2. Calculate percentages:—
(a) Ore taken, 1 gram. Solution used, 65.2 c.c. Standard, 0.987 gram.
[Pg 74]
(b) Ore taken, 1 gram. Barium sulphate got, 1.432 gram. Barium
sulphate contains 13.73 per cent. of sulphur, and the percentage
of sulphur in the ore is wanted.
(c) Barium sulphate is BaSO4. Calculate the percentage of sulphur
it contains, for use in the preceding question.
3. A method of estimating the quantity of peroxide in a manganese ore
is based on the following reactions:—
(1) MnO2 + 4HCl = MnCl2 + Cl2 + 2H2O.
(2) Cl + KI = KCl + I.
To how much MnO2 is 1 gram of Iodine (I) equivalent?
4. A mineral has the following composition:—
Carbonic acid (CO2) 19.09
Copper oxide (CuO) 71.46
Water (H2O) 9.02
What is its formula?
5. How much copper is contained in 1.5 gram of crystallized copper sulphate
(CuSO4.5H2O)? How much of these crystals must be taken
to give 0.4 gram of copper?
6. How much ferrous sulphate crystals (FeSO4.7H2O) must be taken to
yield 2 litres of a solution, 100 c.c. of which shall contain 0.56
gram of iron?
7. Galena is PbS, and hæmatite Fe2O3. What percentages of metal do
these minerals contain?
The relation of the weight of a substance to its volume should be kept in mind in all cases where both weight and volume are dealt with. Students are apt to imagine that on mixing equal volumes of, say, sulphuric acid and water, an acid of half the strength must be obtained. If the statement of strength is in parts by weight this will lead to considerable error. For example, 100 c.c. of sulphuric acid containing 98 per cent. by weight of real acid, will, if diluted with 100 c.c. of water, yield a solution containing not 49 per cent. by weight, but about 63.5 per cent. of the acid. The reason is this: the 100 c.c. of sulphuric acid weighs 184 grams, and contains 180.32 grams of real acid, while the 100 c.c. of water weighs only 100 grams; the mixed water and acid weighs 284 grams, and contains 180.32 of real acid, which is equivalent to nearly 63.5 per cent. by weight. If, however, the method of statement be volumetric, it would be correct to say that doubling the volume halves the strength: if 100 c.c. of brine contains 10 grams of salt, and is diluted with water to 200 c.c., it would be of one-half the former strength, that is, 100 c.c. of the solution would contain 5 grams of salt.
This confusion is avoided by always stating the strengths as so many grams or "c.c." in 100 c.c. of the liquid. But obviously it would be advantageous to be able to determine quickly the weight of any particular substance corresponding to 1 c.c. or some other given volume. Moreover, in descriptions of processes the strengths of acids and solutions are frequently defined neither by their gravimetric nor volumetric composition, but by a statement either of specific gravity or of the degrees registered by Twaddell's or Beaumé's hydrometer. Thus, in the description of the process of gold parting, one writer gives: "The acid should be of 1.2 specific gravity"; and another says: "The acid must not be stronger than 32° Beaumé."
These considerations justify an account of the subject in such a work as this. And on other grounds the determination of a specific[Pg 76] gravity is one of the operations with which an assayer should be familiar.
The meaning of "specific gravity" is present in the mind of every one who uses the sentence "lead is heavier than water." This is meaningless except some such phrase as "bulk for bulk" be added. Make the sentence quantitative by saying: "bulk for bulk lead is 11.36 times heavier than water," and one has the exact meaning of: "the specific gravity of lead is 11.36." A table of the specific gravities of liquids and solids shows how many times heavier the substances are than water.
It is better, however, to look upon the specific gravity (written shortly, sp. g.) as the weight of a substance divided by its volume. In the metric system, 1 c.c. of water at 4° C. weighs with sufficient exactness 1 gram; consequently, the sp. g., which states how many times heavier than water the substance is, also expresses the weight in grams of one c.c. of it. So that if a 100 c.c. flask of nitric acid weighs, after the weight of the flask has been deducted, 120 grams, 1 c.c. of the acid weighs 1.2 gram, and the sp. g. is 1.2. The specific gravity, then, may be determined by dividing the weight of a substance in grams by its volume in c.c.; but it is more convenient in practice to determine it by dividing the weight of the substance by the weight of an equal volume of water. And since the volumes of all substances, water included, vary with the temperature, the temperature at which the sp. g. is determined should be recorded. Even then there is room for ambiguity to the extent that such a statement as the following, "the specific gravity of the substance at 50° C. is 0.9010," may mean when compared with water at 50° C. or 4° C., or even 15.5° C. For practical purposes it should mean the first of these, for in the actual experiments the water and the substance are compared at the same temperature, and it is well to give the statement of results without any superfluous calculation. In the metric system the standard temperature is 4° C., for it is at this point that 1 c.c. of water weighs exactly 1 gram. In England, the standard temperature is 60° F. (15.5° C.), which is supposed to be an average temperature of the balance-room. The convenience of the English standard, however, is merely apparent; it demands warming sometimes and sometimes cooling. For most purposes it is more convenient to select a temperature sufficiently high to avoid the necessity of cooling at any time. Warming to the required temperature gives very little trouble.
Determination of Specific Gravity.—There is a quick and easy method of determining the density or sp. g. of a liquid, based upon the fact that a floating body is buoyed up more by a heavy liquid than by a light one. The method is more remarkable for[Pg 77] speed than accuracy, but still is sufficiently exact. The piece of apparatus used for the purpose is endowed with a variety of names—sp. g. spindle, hydrometer, areometer, salimeter, alcoholimeter, lactometer, and so on, according to the special liquid upon which it is intended to be used. It consists of a float with a sinker at one end and a graduated tube or rod at the other. It is made of metal or glass. Generally two are required, one for liquids ranging in sp. g. from 1.000 to 2.000, and another, which will indicate a sp. g. between 0.700 and 1.000. The range depends on the size of the instrument. For special work, in which variations within narrow limits are to be determined, more delicate instruments with a narrower range are made.
In using a hydrometer, the liquid to be tested is placed in a cylinder (fig. 34) tall enough to allow the instrument to float, and not too narrow. The temperature is taken, and the hydrometer is immersed in the fluid. The mark on the hydrometer stem, level with the surface of the liquid, is read off. With transparent liquids it is best to read the mark under and over the water surface and take the mean.
The graduation of hydrometers is not made to any uniform system. Those marked in degrees Baumé or Twaddell, or according to specific gravity, are most commonly used. The degrees on Baumé's hydrometer agree among themselves in being at equal distances along the stem; but they are proportional neither to the specific gravity, nor to the percentage of salt in the solution. They may be converted into an ordinary statement of specific gravity by the following formulæ:—
Sp. g. = 144.3/(144.3-degrees Baumé.)
or putting the rule in words, subtract the degrees Baumé from 144.3, and divide 144.3 with the number thus obtained. For example: 32° Baumé equals a sp. g. of 1.285.
144.3/(144.3-32) = 144.3/(112.3) = 1.285
This rule is for liquids heavier than water; for the lighter liquids the rule is as follows:—
Sp. g. = 146/(136 + degrees Baumé.)
or in words divide 146 by the number of degrees Baumé added[Pg 78] to 136. For example: ammonia of 30° Beaumé has a sp. g. of 0.880 (nearly).
146/(136+30) = 146/166 = 0.8795
A simple series of calculations enables one to convert a Beaumé hydrometer into one showing the actual sp. g. Graduation, according to sp. g. is the most convenient for general purposes. In these instruments the distances between the divisions become less as the densities increase.
Twaddell's hydrometer is graduated in this way: Each degree Twaddell is 0.005 in excess of unity. To convert into sp. g. multiply the degrees Twaddell by 0.005, and add 1. For example: 25° Twaddell equals a sp. g. of 1.125.
25×.005 = 0.125; + 1.000 = 1.125.
There is a practice which ignores the decimal point and speaks of a sp. g. of 1125 instead of 1.125. In some cases it is convenient, and inasmuch as no substance has a real sp. g. of much over 20, it can lead to no confusion. The figures expressed in this way represent the weight of a litre in grams.
Some hydrometers are graduated so as to show at a glance the percentage composition of the liquid they are intended to be used with. Gay-Lussac designed one to show the alcoholic strength of mixtures of alcohol and water; the construction of others upon the same principle is easy and perhaps useful. But when the principle is applied to complex liquids and mixed solutions, it is misleading.
The various methods of graduation ought all to give place to one showing a simple statement of the sp. g.
The method of determining sp. g. with the hydrometer is obviously inapplicable to the case of solids, and in the case of liquids it should not be used where exact figures are required. There are several other methods which may be used, but on the whole those with the specific gravity bottle are most convenient.
The specific gravity bottle (fig. 35) is a light flask of about 25 c.c. capacity, provided with a well-fitting perforated stopper. It is essentially a graduated flask, which measures a constant volume, but it does not much matter what the volume is.
In taking the sp. g. of a liquid (or, what is the same thing, a fused solid) there is wanted the weights (1) of the flaskful of water and (2) of the flaskful of the liquid. Dividing the second by the first gives the required sp. g. The actual weighings required are[Pg 79]—
(1) of the dry and empty flask,
(2) of the flask filled with water, and
(3) of the flask filled with the liquid.
The weighing of the flask once made need not be often repeated. It is well to do so now and then for safety's sake; but one weighing will serve for a large number of determinations. The same remarks apply to the weighing of the bottle filled with water. The bottle is dried by rinsing out first with alcohol and afterwards with ether; ether is very volatile, and a short exposure in a warm place will soon drive off the little remaining about the sides. The ether vapour should be sucked out through a glass tube. See that the bore of the stopper is dry as well as the bottle. Let the dry bottle stand in the box of the balance for a minute or two before weighing. The weight is, strictly speaking, not that of the empty bottle, but of the bottle filled with air. The empty bottle would weigh from 20 to 30 milligrams less. Correcting for this would, in most cases, only make a difference in the fourth place of decimals,[8] so that it is better to ignore the error.
The weight of the flask filled with water is got by filling it with distilled water, and inserting the stopper. The excess of water will overflow at the margin and through the bore. The bottle is wiped with a soft, dry cloth, taking care not to squeeze or warm the bottle. The bottle will remain filled to the top of the stopper. It is allowed to stand in the balance box for a minute or two, and then weighed.
Distilled water, as stated, should be used; the use of ordinary water may increase the weight by 5 or 6 milligrams. Many waters, if they have not previously been boiled, give off bubbles of air which render the weighing worthless.
The temperature of the water is of greater importance; lowering the temperature 2° will increase the weight by 10 or 12 milligrams. A beaker of water may be warmed or cooled to the required temperature; then the bottle is filled from it, and quickly weighed. If the balance-room is cooler than the water, the latter will draw back into the bottle, and a few small bubbles of air will enter; but even in extreme cases this will only increase the weight by a very small fraction of a milligram. There is more trouble caused when the room is warmer, for the liquid then expands and protrudes as a drop resting on the top of the stopper.[Pg 80] There will in this case be loss by evaporation, which in the case of the more volatile liquids, such as alcohol, is serious. To prevent this loss, as well as any that may arise by overflow, the stopper should be dilated above into a small cup, A (fig. 36), which may itself be stoppered. In a bottle of this kind the neck of the stopper is graduated, and the bottle is considered full when the liquid stands at the level of the mark in the neck. On inserting the stopper, the liquid rises into the cup, and is reduced to the level of the mark by absorption with pieces of filter-paper.
For most purposes, however, there is no need for cooling and allowing room for subsequent expansion. The assayer, as a rule, can select his own standard temperature, and may choose one which will always necessitate warming. It will be handier in this case to have a bottle with a thermometer stopper. Of the two types shown in fig. 37, that with the external thermometer tube (A) is more generally useful.
The bottle is filled at a lower temperature, and is then gently warmed so as to slowly raise the temperature to the required degree. The superfluous liquid is then at once wiped off, and the bottle cooled and weighed.
The weight of the flask filled with the liquid whose sp. g. has to be determined is ascertained in a similar way. Of course the temperature must be the same. If the liquid does not mix with water, the bottle should be dried before filling, but otherwise the flask need only be rinsed out two or three times with the liquid.
Having obtained the three weighings, deduct the weight of the bottle from each of the others to get the weights of the water and liquid respectively. Divide the latter by the former, the result shows the sp. g. As an example, take the following, in which a rather large sp. g. bottle was used:—
| 1. Weight of bottle | 39.299 | gram |
| 2. Weight of bottle and water | 81.884 | " |
| 3. Weight of bottle and paraffin | 73.146 | " |
By subtracting 1 from 2 and 3 the result is as follows:[Pg 81]—
| 81.884 | grams | 73.146 | grams |
| 39.299 | " | 39.299 | " |
| —————— | —————— | ||
| 42.585 | of water. | 33.847 | of paraffin. |
Divide the weight of the paraffin by that of the water—
42.585)33.8470(0.7948
29.8095
——————
.......
The sp. g. of the paraffin is 0.7948.
The sp. g. of a fusible solid may be obtained in the same way at a temperature some degrees above its fusing point.
The sp. g. of a solid in powder or gravel sufficiently fine to pass through the neck of the bottle is easily determined. If the bottle filled with water weighs 50 grams, and there is placed on the pan alongside of it 20 grams of a sand, the weight of the two together will of course be 70 grams. But if the sand is put in the bottle, it evidently displaces its own bulk of water; and if, on again weighing, the weight is found to be 62 instead of 70 grams, it is because the 20 grams of sand has displaced 8 grams of water. Bulk for bulk, the sand is 2-1/2 times as heavy.
In practice, the weight of the bottle filled with water will probably be already known; if not, it must be determined. A certain quantity, say 20 grams, of the powdered substance is then transferred carefully to the bottle. The bottle need not be dry inside, but its neck and outside must be. In making this transference a careful worker will make no loss, and the mode of working saves a little time. But it is better to weigh the dry flask; put into it 10 to 20 grams of the powder, and weigh again. The increase in weight gives accurately the weight of powder in the bottle. About two-thirds fill the bottle with distilled water, and mix with the powder by gentle shaking. Air bubbles will disentangle themselves, and rise to the surface of the water. Wash back anything adhering to the stopper with a jet of water, and fill the bottle almost to overflowing. Allow it to stand for a minute or so; replace the stopper; warm to the required temperature; take off the superfluous moisture; wipe and weigh. As an example, take the following:—
| 1. | Weight | of | bottle | 12.681 | grams |
| 2. | " | " | bottle filled with water | 37.708 | " |
| 3. | " | " | bottle with wolfram | 40.821 | " |
| 4. | " | " | bottle with wolfram and water | 61.199 | " |
Subtract (1) from (3) to get the weight of wolfram taken:
40.821 grams
12.681 "
——————
28.140 "
add the weight of the wolfram to the weight of the bottle filled with water:
28.140 grams
37.708 "
——————
65.848 "
subtract (4) from this to get the weight of water displaced:
65.848 grams
61.199 "
——————
4.649 "
Divide the weight of the wolfram by the weight of the water displaced to get sp. g.:
4.649)28.140(6.053
27.894
———————
......
If the solid is soluble in water, or has a tendency to float, some liquid other than water is used. Paraffin oil or oil of turpentine will do. The process is as follows:—The weight of the dry and empty bottle having been determined, add a sufficiency of the substance and weigh again to find how much has been added. Fill up with paraffin oil and weigh again. Clean out the substance by rinsing with paraffin; fill up and weigh. Calculate the sp. g. as if water had been used, and multiply by the sp. g. of the paraffin.
For example:
| 1. | Weight | of | bottle | 39.299 | grams |
| 2. | " | " | bottle and nitre | 57.830 | " |
| 3. | " | " | bottle and paraffin | 73.146 | " |
| 4. | " | " | bottle and paraffin and nitre | 84.665 | " |
| 5. | " | " | bottle and water | 81.884 | " |
First from (1),(3), and (5), calculate the sp. g. of the paraffin as already shown. It will be 0.7948. Deduct (1) from (2) to get the weight of the nitre:
57.830 grams
39.299 "
——————
18.531 "
[Pg 83]
add this to (3):
18.531 grams
73.146 "
——————
91.677 "
and deduct (4) to find the weight of the equal bulk of paraffin.
91.677 grams
84.665 "
——————
7.012 "
divide the weight of the nitre by the weight of the paraffin:
7.012)18.531(2.6427
——————
......
The sp. g., taking paraffin as the standard instead of water, is 2.6427. Multiply this by the sp. g. of paraffin, 0.7948, and the result is 2.1004 as the sp. g. of nitre compared with water.
Similarly, a sp. g. compared with water at say 50° C. can be converted into one compared with water at standard temperature, by multiplying by the sp. g. of water at 50° C. The following table gives the sp. g. of water at various temperatures:—
| Degrees Centigrade. | Sp. G. | Degrees Centigrade. | Sp. G. | Degrees Centigrade. | Sp. G. |
| 4° | 1.0000 | 20° | 0.9982 | 40° | 0.9923 |
| 10° | 0.9997 | 25° | 0.9971 | 50° | 0.9881 |
| 15° | 0.9991 | 30° | 0.9957 | 100° | 0.9586 |
If, for example, a substance at 50° C. has a sp. g. of 0.9010 as compared with water at 50° C., it will have (compared with water at 4° C.) a sp. g. of 0.9010 × 0.9881; or 0.8903. The figures 0.8903 represent the sp. g. of the substance at 50° C. compared with water at 4° C. Except in comparing the sp. gravities of the same substance at different temperatures, a calculation of this kind serves no useful purpose.
In taking the specific gravity of a solid not in powder, a lump of it is freed from loose particles and its exact weight determined. By means of a horse hair with a slip knot it is suspended to the balance, and beneath it is placed, out of contact with the balance pan, a beaker of distilled water. The horse hair must be long enough to keep the mineral well beneath the surface of the water so as to allow the balance to vibrate. Air bubbles are removed by touching with a camel-hair pencil. Whilst the mineral is suspended in water the weight is again taken. It will weigh less than before, and the difference between the two weighings gives[Pg 84] the weight of water (and consequently the volume) displaced by the mineral. The weight in air divided by the difference is the specific gravity. Thus
Weight in air 3.2170 grams
Weight in water 2.7050 "
———
Difference 0.5120 gram
3.2170/0.5120 equals 6.28, the sp. g.
The sp. g. of a substance depends mainly on its composition, but is affected by certain conditions. The effect of temperature has been already considered. Air holes and empty spaces lessen the specific gravity of otherwise solid bodies; and metals, which after fusion become imperfect solids, have their density increased by hammering or rolling. But metals when free from pores have their density diminished when rolled, without annealing. The effects of these conditions are slight when compared with those due to the presence of impurities.
For simple substances, or mixtures of only two substances, a determination of sp. g. is a sufficient check on the composition for many practical purposes; and with more complex mixtures, such as slags and some of the products of dressing operations in which the material does not differ much in its nature from time to time, such a determination will yield information of considerable value, and afford a check upon the proper working of a process.
When the mixing of two substances is accompanied by a change in volume, the sp. g. of the mixture can only be learnt by experiment. But when the substances have no such action on each other the resulting sp. g. can be calculated. Some of these calculations have a practical interest as well as an educational value. Students should practise them so as to become familiar with the relations between weight and volume.
When substances are mixed by volume, the sp. g. of the mixture is the mean of those of its constituents, and may be calculated in the usual way for obtaining averages. 1 c.c. of a substance having a sp. g. of 1.4 mixed with 1 c.c. of another having a sp. g. of 1.0 will yield 2 c.c. of a substance having a sp. g. of 1.2. If, however, we write gram instead of c.c. in the above statement, the resulting sp. g. will be 1.16. The simplest plan is to remember that the sp. g. is the weight divided by the volume (sp. g. = w/v) and the sp. g. of a mixture is the sum of the weights divided by the sum of the volumes (sp. g. = (w + w' + w", &c.)/(v + v' + v", &c.)). In the above example the sum of the volumes is 2 c.c.; the weights (got by multiplying each volume by its[Pg 85] corresponding sp. g.) are 1.4 gram and 1 gram. The sum of the weights divided by the sum of the volumes is 2.4/2 or 1.2.
The sp. g. of a mixture of 10 c.c. of a substance having a sp. g. of 1.2, with 15 c.c. of another having a sp. g. of 1.5 may be thus found:—
sp. g. = (12+22.5)/(10+15) = 1.38
multiply each volume by its sp. g. to get its weight:
10×1.2 = 12 15×1.5 = 22.5
add these together (12+22.5 = 34.5) and divide by the sum of the volumes (10+15 = 25):
25)34.5(1.38
25
—
95, &c.
The sp. g. will be 1.38, provided the mixture is not accompanied by any change of volume.
The same formula will serve when the proportion of the ingredients is given by weight. A mixture of 4 parts by weight of galena (sp. g. 7.5) with 5 parts of blende (sp. g. 4) will have a sp. g. of 5.06:
sp. g. = (4+5)/(0.53+1.25) = 9/1.78 = 5.06
It is necessary in this case to calculate the volumes of the galena and of the blende, which is done by dividing the weights by the sp. gravities: thus, 4 divided by 7.5 gives 0.53 and 5 divided by 4 gives 1.25.
The converse problem is a little more difficult. Given the sp. g. of a mixture and of each of the two ingredients, the percentage by weight of the heavier ingredient may be ascertained by the following rule, which is best expressed as a formula. There are three sp. gravities given; if the highest be written H, the lowest L and that of the mixture M, then:
Percentage of heavier mineral = (100×H×(M-L))/(M×(H-L))
Suppose a sample of tailings has a sp. g. of 3.0, and is made up of quartz (sp. g. 2.6) and pyrites (sp. g. 5.1): then the percentage of pyrites is 27:
(100×5.1×(3-2.6))/(3×(5.1-2.6)) = (510×0.4)/(3×2.5) = 204/7.5 = 27.2
The same problem could be solved with the help of a little algebra by the rule already given, as thus: the sp. g. of a mixture[Pg 86] equals the sum of the weights of the constituents divided by the sum of the volumes. Then 100 grams of the tailings with x per cent. of pyrites contain 100-x per cent. of quartz. The sum of the weights is 100. The volume of the pyrites is x/5.1 and of the quartz (100-x)/2.6.
Then we have by the rule
3 = 100/((x/5.1)+(100-x)/2.6)
3 = 1326/(510-2.5x)
204 = 7.5x
and x = 27.2
If the percentage (P) and sp. g. (H) of one constituent and the sp. g. (M) of the mixture are known, the sp. g. of the other constituent may be calculated by the following formula, in which x is the required sp. g.:
x = ((100-P)×M×H)/((100×H)-(P×M))
For example, "tailings" (sp. g. 3.0) containing 27.2 per cent. of pyrites (sp. g. 5.1) will contain (100-27.2), 72.8 per cent. of earthy matter having a mean sp. g. of x:
x = ((100-27.2)×3×5.1)/((100×5.1)-(27.2×3))
= 1113.84/428.4 = 2.6
The differences in sp. g. corresponding to differences in strength have been carefully determined and tabulated in the case of the stronger acids and of many other liquids. Such tables are given at the end of this book.
To Calculate the Weight of a Measured Volume of Mineral or Rock.—Multiply the cubic feet by 62.4 and then multiply by the sp. g. of the stuff, the answer gives the weight in pounds. For example, 100 cubic feet of quartz weighs 100×62.4×2.6 = 16,224 lbs. The weight of any mass of mineral of known extent and sp. g. is ascertained in this way.
The following table gives the specific gravities of some of the commoner minerals.
| Barytes | 4.5 |
| Blende | 4.0 |
| Calcite | 2.6 |
| Cassiterite | 6.9 |
| Chalybite | 3.8 |
| Copper pyrites | 4.2 |
| Fluor | 3.1 |
| Galena | 7.5 |
| Hæmatite | 5.0 |
| Mispickel | 6.2 |
| Pyrites | 5.0 |
| Quartz | 2.6 |
[8] The difference of 20 or 30 milligrams is disregarded here because it detracts equally from the actual weight of the water and liquid to be determined. If the liquid is a heavy one the difference shows itself in the third or second place of decimals. The correction may be made by deducting from the weight of the flask 0.0012 grams for each gram of water it holds.
Silver is widely diffused, and has been found in most mining districts. It occurs native in sufficient quantity to constitute one of the chief ores of the metal. It also occurs combined with sulphur (as in argentite), with sulphur and antimony (as in stephanite or brittle silver ore, and in pyrargyrite or ruby silver), and with copper, sulphur, antimony, and arsenic, as in polybasite. Chloride of silver occurs native as horn silver or kerargyrite. Silver is found in the ores of other metals, such as fahlerz, which sometimes contains from two to ten per cent. of the metal, and galena, which is an important source of it; in fact, galena is never found entirely free from silver. It is present also in greater or less quantity in the ores of copper and zinc.
Silver dissolves readily in nitric acid, forming silver nitrate. It only forms one family of salts, and of these the chloride and nitrate are of chief importance to the assayer. The formation of the chloride of silver on the addition of hydrochloric acid or a soluble chloride to the nitric acid solution, serves for the recognition and separation of silver. The precipitated chloride is white (becoming violet on exposure to light), insoluble in nitric acid, soluble in ammonia, hyposulphite of soda, or concentrated solutions of chlorides. The best confirmatory test is made by wrapping the precipitate in a little sheet lead, and cupelling, when the silver will be left in the metallic state, and is easily recognized.
Dry Assay.—This assay is made up of two parts: (1) the concentration of the silver in a button of lead; and (2) the cupellation of the resulting alloy. The concentration of the[Pg 88] button of lead may be effected either by scorification or by fusion in a crucible.
The scorification assay is performed in a scorifier, which is a shallow open-mouthed dish about 2-1/2 inches across, with a very thick bottom to enable it to withstand the corrosive action of the slag. A charge of more than 3 or 5 grams of the ore cannot be worked in one, and with such small charges the unavoidable variations have a serious effect on the figures reported. A difference of one milligram on the weight of the button of silver got represents a difference of 6 or 10 ounces per ton. With rich ores such variation is unavoidable under any conditions, and the only safe plan is to take the mean of several assays. But with poorer ores the accuracy of the assay, as well as convenience in working, is much increased by working in a crucible with larger charges.
In scorification the proportion of lead required for scorifying 1 gram of ore is in average cases from 10 to 15 grams, sinking in the case of galena to 2 grams, and rising with earthy and refractory substances to from 30 to 40 grams. But by fusing in a crucible with well-selected fluxes, a proportion of 4 of flux to 1 of ore is generally sufficient; and not only is the proportion of added matter less, but it is also easier to manipulate large quantities in crucibles, so that, although in some cases the crucible assay is more troublesome and less satisfactory, yet with poor and earthy ores it is the best method of dealing with them; while when properly worked it yields results as accurate as scorification does. As a general rule, if more than 5 grams of ore must be taken, the crucible assay should be adopted.
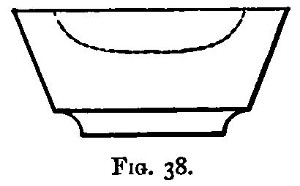
Scorification Assay.—The charge of ore is usually 3 grams, sometimes 5; the lead varies from 30 to 70 grams, and the quantity of soda, borax, or powdered glass added varies from 0.3 to 3 or 4 grams. It is generally recommended to have the lead granulated,[9] and to mix the ore with about half of it in the scorifier; then to put on the rest of the lead; and finally to sprinkle the borax or glass on the top. It answers just as well, however, to use the lead in the shape of foil, and wrap the ore up in it; and if the ore contains much sulphur, the borax may with advantage be added (wrapped in a little tissue paper) some five or ten minutes after the operation has started.[Pg 89]
The process of scorification is as follows:—A scorifier (fig. 38) of convenient size having been selected (one 2-1/2 inches across is most generally useful), it is dried at a gentle heat for about ten minutes. The charge is then put into it, and it is introduced, with the help of a scorifier tongs (fig. 39), into a muffle heated considerably above redness. The muffle is then closed, and when the metal has melted down, it is opened, but the temperature is kept up. A ring of slag will, after a time, form around the metal, and when this appearance (known as the eye) presents itself, the temperature may be lowered. When the eye has disappeared—that is, when the layer of slag has quite closed in—a pinch of powdered culm wrapped in tissue paper is added. As soon as the slag has again become tranquil, the scorifier is taken out, and its contents are poured into a mould (fig. 40), the slag is detached, and saved. If the button of metal weighs more than 30 grams, its size is reduced by another scorification in the same scorifier, which should have been replaced in the muffle immediately after the contents had been poured out. If the ore is not a very rich one, the button of lead will carry practically all the silver; but with rich ores it is more satisfactory to save the slag, and subsequently to melt it down with the cupel on which the lead has been treated, so as to recover the silver lost in the slag, together with that absorbed in the cupel, at one operation. Or, if the cupellation loss is neglected or calculated in some other manner, the slag or slags from the scorifier may be powdered and mixed with 20 grams of oxide of lead, 5 grams of borax, and 1 gram of charcoal. This should be melted down in a small crucible, and the resulting button of lead cupelled.
If the scorification has been unsatisfactory, the quantity of silver obtained from the slag will be by no means inconsiderable. The usual explanation is that with sulphury ores compounds of metallic oxides and sulphides (oxysulphides) are formed, which remain in the slag, retaining considerable quantities of the precious metal. It is said that under certain conditions such a slag may contain as much as 10 per cent. of silver. An excess of lead and a high temperature prevents the formation of these oxysulphides. But if much silver is present in the ore, the slag cannot be safely[Pg 90] thrown away, even if sulphur is absent, and the process has been satisfactorily performed.
If the crust which appears on the surface of the lead does not clear, add a small lump of borax and 20 grams more lead; then close the muffle, and keep the temperature as high as possible. If the slag forms properly, but shows unfused or only half-fused lumps, even when the scorification has proceeded for some time, add more borax, and stir with an iron rod. The slag adhering to the rod must be detached by hammering, and replaced in the scorifier.
If the ore consists largely of quartz, soda should be added instead of borax; or, if it contains much copper, powdered quartz may be used. If the scorifier at the end of an operation is more than usually corroded, the borax should be replaced in subsequent assays on similar ores by powdered glass or quartz.
If a fairly fluid slag is formed which does not clear from the metal and show the eye, more lead and a higher temperature is wanted.
As a general rule, it may be stated that when a scorification is unsatisfactory, what is wanted is more heat, more lead, or more borax.
It is a safe plan when work has to be done on a strange ore, to make three or four assays with varying quantities of lead. The proportion of lead is right when a further addition does not yield a higher result. The proper proportion having been found, a note of it should be made for future use.
The object of the fusion in a crucible, like that of scorification, is to concentrate the silver in a button of lead which is to be subsequently cupelled; and to retain the earthy and waste matters in the slag. It is necessary to consider the quality of the slag and the weight and quality of the lead. The slag when fused should be liquid and homogeneous, and not too corrosive on the crucible. The button of lead should be soft, malleable, and free from a coating of regulus.[10] In weight it should not differ much from the ore taken. With 20 grams of ore, for example, a button of lead weighing from 18 to 25 grams will be satisfactory: less than this would leave an undue proportion of silver in the slag; and more would be unnecessarily large for cupelling, and would increase the loss in that operation.
With average ores, take 20 grams of the powdered ore and mix with 30 grams of "soda," 40 grams of red-lead or litharge, 5 grams of borax, and from 2 to 2.5 grams of flour, and place in an E crucible[Pg 91] (Battersea round). Put these in the furnace at a red heat, cover the crucible, and gradually raise the temperature until the whole charge has melted down and is in a state of tranquil fusion. Pour into a mould, and replace the crucible in the furnace. As soon as the lead is solid, detach the slag and put it back into the crucible; and when it is again fluid, charge on to it with a copper scoop a mixture of 20 grams of oxide of lead, and 1 gram of charcoal: when fusion has again become tranquil, pour and detach the button of lead. The lead buttons should be hammered into discs with rounded edges, and be freed from slag; if too big for a cupel they may be scorified together in a small scorifier, but it is better to cupel them separately.
Ores containing Metallic Oxides.—Peroxides of iron, manganese, and copper interfere by counteracting the effect of the charcoal or flour, and thus reducing the size of the lead button. Peroxide of iron will reduce the weight of lead by a little more than its own weight; and peroxide of manganese has about twice this effect. When these oxides are present an additional quantity of flour must be used, and precautions must be taken to prevent reoxidation of the slag by the furnace gases. This may best be prevented by using a layer of common salt as a cover to the charge. When the ores contain a good deal of quartz or stony matter, the fluxes just given (for average ores) will do; but the proportion of soda should be diminished, and that of the borax, oxide of lead, and flour increased as the quantity of metallic oxides become greater. If the ore contains practically no quartz, the soda may be altogether omitted, and some glass or powdered quartz added. The following charge may be taken as an example: weigh up 20 grams of the powdered ore, 15 grams each of "soda" and borax, 60 grams of oxide of lead, and 5 grams of flour. Mix and place them in an E crucible, and cover with a layer of from a quarter to half an inch of common salt. Place in the furnace as before. The salt will give off a considerable amount of fume, which will, to a certain extent, conceal the state of the charge: when the crucible has been in the furnace for about 25 minutes remove it and pour out the contents immediately. With ores that produce a thick slag the addition of 5 grams of fluor spar will be an advantage. It may happen that with an unknown ore the first assay will be more or less unsatisfactory: but from it the necessity for adding more or less flour will be learnt, and a second assay, with the necessary modification of the charge, should give a good result.
Ores containing much Sulphides.—Ores of this class may be easily recognized, either by the appearance of the minerals they contain or by the odour of sulphurous oxide (SO2) which they evolve when roasted on a spatula. The sulphides most commonly[Pg 92] present, in addition to the sulphurized minerals of silver, are pyrites, galena, blende, and mispickel. When they are present in only a moderate amount, their effect is simply to increase the weight of the button of lead; and this is easily counteracted by reducing the amount of flour, or by omitting it. When in larger amounts, they not only yield large buttons, but also render the metal sulphury, sometimes even giving a button of regulus instead of lead. This last evil may be remedied (1) by putting in a rod of iron as soon as the charge has fused, or (2) it may be counteracted by a proper addition of nitre, or (3) when the sulphides present are only those of iron or copper the sulphur may be removed by calcining, and the ore converted into one of the class containing metallic oxides. The calcination is effected as follows:—Weigh up 20 grams of the powdered ore and place it in a wide-mouthed crucible sufficiently large to perform the subsequent melting down in. The roasting must be done at a gentle heat at first, so as to avoid clotting: the mouth of the crucible should project considerably above the coke, and should slope forward towards the worker. The charge must be occasionally stirred with the stirrer (fig. 10) so as to expose fresh surfaces to the action of the air, and to prevent adhesion to the sides of the crucible. The stirrer should not be removed till the calcination is finished. The temperature should be raised at the end to a good red heat; and (to ensure the decomposition of any sulphate that may be formed) the roasted ore should be rubbed up in a mortar with a pinch of anthracite, and again calcined. It is then mixed with fluxes as described, and fused in the same crucible.
The calcination of an ore is a work occupying a good deal of time, and, in most cases, it is better to take advantage of the desulphurizing power of red lead or nitre. Red lead by itself will do, but a large quantity of it will be required; 1 part of a metallic sulphide needs from 20 to 50 parts of red lead to yield a button free from sulphur; whereas at most from 2 to 2-1/2 parts of nitre are sufficient. There is sometimes an advantage in having a considerable excess of oxide of lead in the slag, but where there is no such reason, 2 parts of red lead to 1 of ore is enough. A charge which will do for most sulphides is the following: 20 grams of ore, 40 to 100 grams of red lead, 20 grams of "soda," 5 of borax, and sufficient nitre (or perhaps flour) to give a button of about 25 grams of lead. How much this must be (if not already known) may be approximately determined by fusing 3 grams of the ore and 3 grams of "soda" in a small crucible (C) with 50 grams of litharge (not red lead) under a cover of salt, and weighing the resulting button of lead. Subtract 3 from the weight of lead obtained, and the difference multiplied by 1.3 will give the quantity in[Pg 93] grams of nitre required. If the button of lead weighs less than 3 grams flour must be added. If this is not satisfactory repeat the assay, adding an extra gram of nitre for each 4 grams of lead in excess of that required, or 1 gram of flour for a 12-gram deficiency.
In the method in which iron is used as a de-sulphurising agent, only as much oxide of lead should be added as will give a button of lead of the required size. Rather a large button of lead should be got, and the slag should be strongly alkaline; if the ore does not already carry a large amount of sulphur some should be added. The fusion should be performed at a low temperature (similar to that for a galena assay), and should be continued for some time after it has become tranquil. Take 20 grams of the ore, 40 grams of "soda," 40 grams of oxide of lead, and 5 or 10 grams of borax; place this mixture in a crucible (with a rod of iron, as in the galena assay), cover, and fuse for about half an hour. Take out the rod, washing it in the slag, and, in a minute or two, pour. Clean and cupel the button of lead.
General Remarks on the Fusion.—Other things being equal, the smaller the quantity of the slag the better, provided there is sufficient to cover the metal. The presence of peroxides of the heavy metals is prejudicial, since they tend to increase the quantity of silver retained in the slag. It may be given as a general rule that when iron, copper, manganese, &c., are present, there is a more than ordinary need for cleaning the slags, and care must be taken to keep these metals in the state of lower oxide.
In selecting the fluxes, it should be remembered that soda is the best for quartz, and borax for lime and metallic oxides. And that with ores almost free from gangue some quartz or glass should be added to protect the crucible. Two parts of soda are enough to flux 1 part of quartz; whilst of borax, or oxide of lead, 4 parts are barely sufficient. Oxide of lead has the advantage of being heavy and so does not occupy much space in the crucible; on the other hand, if the melting down be performed too quickly, or if oxide of lead only is used, this high specific gravity is a disadvantage, for the lighter earthy matter floats as a pasty mass on the more fluid oxide of lead, and thus escapes its action.
When metallic sulphides are present in the ore, an excess of oxide of lead helps to keep the sulphur out of the button of metal. In addition to the oxide of lead required as a flux, some will be required to provide the lead in which the silver is to be collected. Oxide of lead, mixed with charcoal or flour, yields, when heated, a multitude of minute buttons of metal uniformly distributed through the mass of the charge; as the charge melts down these run together and fall to the bottom; this shower of lead collects[Pg 94] the silver more easily than a single button at the bottom of the crucible could do. Only that portion of the oxide of lead which remains in the slag can be considered as a flux; very often the first indication of an excessive reduction of lead is the pastiness of the slag rendered thick by the withdrawal of the oxide of lead which would have kept it fluid. If, in an assay, it is found that 5 parts of flux are not sufficient for 1 part of ore, the remedy lies in using a different flux rather than in taking a larger quantity.
On the Reducing Effect of Charcoal, Flour, and Tartar.—The weight to be got from a given charge will depend (provided sufficient oxide of lead is present) upon the proportion of the reducing agents in it. We have thought it well to illustrate this part of the subject by a series of experiments which the learner will do well to practise for himself before proceeding to the assay of actual ores. Take 80 grams of litharge and 20 grams of a mixture of borax and soda. Fuse three lots (1) with 1.5 gram of charcoal, (2) with 3 grams of flour, and (3) with 7.5 grams of tartar. Weigh the buttons of lead obtained, and divide each by the weight of reducing agent used. The results will differ somewhat with the dryness and quality of the flour, etc., used; in one series of experiments they were as follows:—
| Gram. | Grams. | Gram. | Grams. | |||||||
| 1.5 | charcoal | gave | 34.0 | lead | ∴ | 1 | charcoal | = | 22.6 | lead. |
| 3.0 | flour | " | 33.5 | " | ∴ | 1 | flour | = | 11.2 | " |
| 7.5 | tartar | " | 38.0 | " | ∴ | 1 | tartar | = | 5.0 | " |
The use of flour as a reducing agent has many advantages, and it is well to remember that 1 gram of flour reduces about 11 grams of lead; and that charcoal has twice, and tartar one-half, this reducing effect.
On the Reducing Effect of Charcoal, &c., on Red Lead.—It is often easier to obtain red lead of good quality than it is litharge, and by a large number of assayers red lead is the form of oxide of lead always used. Red lead, however, contains an excess of oxygen which will use up some of the reducing agent before lead separates out. On making a series of experiments (similar to the last, but using 80 grams of red lead instead of the litharge) the results were, with the same quantities of the reducing agents:—
| With | charcoal, | 18 | grams | of lead. |
| " | flour, | 18 | " | " |
| " | tartar, | 22 | " | " |
Comparing these with the results with litharge, in the previous table it will be seen that the same quantity of reducing agent has in each case brought down 16 grams less of lead, so that a larger amount of the reducing agent must be added to get a button of[Pg 95] the same weight as that obtained with litharge. To get a button of a desired weight, say 22 grams, we must add reducing agent sufficient to throw down 22 + 16 or 38 grams of lead, which would require 3.4 grams of flour. If this amount of flour is fused with 80 grams of red lead, a button of lead weighing 22 grams will be formed, the other 16 grams being kept up by the oxygen of the red lead.
If the quantity of red lead differs from 80 grams, this rule must be modified. With 40 grams of red lead, for example, we should add an excess of reducing agent sufficient to throw down 8 grams of lead instead of 16. Similarly, with 160 grams of red lead, we should add enough to throw down 32 grams.
The following rule will enable one to calculate the weight of flour required to produce a button of lead of any desired weight from any given quantity of red lead. Each 5 grams of red lead present diminishes the weight of the lead by 1 gram. If then we divide the weight of red lead in a charge by 5, and add this to the weight of lead required, the sum divided by 11 will give the weight of flour which must be added. Using 80 grams of red lead and wanting a button of 20 grams, we should add 3.3 grams of flour.
80/5 = 16; 16+20 = 36; 36/11 = 3.3 nearly.
The following are some results obtained which will illustrate the rule:—
| Red Lead used. | Flour used. | Lead got. | |||
| 40 | grams | 3 | grams | 25.0 | grams |
| 100 | " | 3 | " | 13.5 | " |
| 80 | " | 4 | " | 30.0 | " |
| 80 | " | 5 | " | 40.0 | " |
On the Reducing Effect of Metallic Sulphides, and the Counteracting Effect of Nitre.—The sulphides found in ores will reduce a button of lead from oxide of lead just as flour does; and, as charcoal, flour and tartar differ in their reducing power, so equal weights of the different mineral sulphides throw down different weights of lead.
One gram of iron pyrites yields about 11 grams of lead. One gram of copper pyrites, blende, fahlerz, or mispickel, yields 7 or 8 grams of lead, whilst 1 gram of antimonite will give 6, and 1 gram of galena only a little over 3 grams. It is evident that if an ore carries much of these sulphides, the quantity of lead reduced will be very much larger than that required for an assay. To counteract this effect nitre is added; 1 gram is added for each 4 grams of lead in excess of that required. For example: with a 20-gram charge of an ore containing 50 per cent. of pyrites, if no nitre were added, 110 grams of lead would be got; or, if there[Pg 96] was not sufficient oxide of lead to yield this quantity of metal, the button would be sulphury. To reduce the weight of the button by 80 grammes, we should add 20 grams of nitre, if litharge were used; or if red lead were used, we should add 16 grams of nitre, since the oxidizing effect of 20 grams of red lead is equivalent to that of 1 of nitre, and since 80 grams of red lead are generally used in a charge. Two assays of an ore of this kind with these quantities of nitre gave 26.0 grams of lead with litharge, and 22.5 grams with red lead.
It is best to use in these assays 80 grams of red lead, 20 of soda, and 5 of borax, with 20 grams of the ore. If the lead got by the preliminary fusion in a small crucible with litharge (described under "ores containing much sulphides") is known, the following table will indicate the quantity of nitre, or flour, to be added with this charge:—
| Lead got in Preliminary Fusion with 3 grams of Ore. | Flour to be added to the Assay. | Nitre to be added to the Assay. |
| 0.0 gram | 3.3 grams | none |
| 3.0 grams | 1.3 gram | — |
| 6.0 " | none | 4.0 grams |
| 9.0 " | — | 9.0 " |
| 12.0 " | — | 14.0 " |
| 15.0 " | — | 19.0 " |
| 18.0 " | — | 24.0 " |
| 21.0 " | — | 29.0 " |
If litharge is used in the assay instead of red lead 4 grams more nitre, or 1.5 gram less flour must be used. When more than a few grams of nitre are added to a charge the proportion of "soda" and borax should be increased, because one of the products of the reaction of nitre upon sulphides in the presence of soda is sulphate of soda, and because the "soda" thus used up no longer serves as a flux; more borax should be added, as it is the best flux for the metallic oxides which are formed in the process. If in an assay too large a button of lead is got, even after this calculation has been made, and the assay is repeated, add 1 gram more nitre for each 4 grams of lead in excess. Sometimes the assay appears tranquil before the nitre has produced its full effect; in such cases it is well to seize the crucible with the tongs and mix its fused contents by rotating them; if this causes an effervescence, the crucible should be replaced in the fire and the fusion continued. The following experiments will illustrate the extent to which the above rules may be relied on. In all of them the standard flux was used, viz.:—80 grams of red lead, 20 of soda, and 5 of borax.[Pg 97]
| Pyrites | 5 | 5 | 5 | 5 | 2.5 | 5 | 10 | 15 | 20 |
| Quartz | — | 20 | — | 20 | 17.5 | 15 | 10 | 5 | |
| Nitre | — | — | 5 | 5 | — | 4 | 16 | 28.5 | 41 |
| Lead got | 42.5 | 36.0 | 16.0 | 19.0 | 11.5 | 22.5 | 22.5 | 26.5 | 27.5 |
| Copper Pyrites | 8 | 8 | 8 | 8 |
| Quartz | — | 12 | — | 12 |
| Nitre | — | — | 4 | 4 |
| Lead got | 47.5 | 34.0 | 33.0 | 26.0 |
| Antimonite | 8 | 8 | 8 | 8 |
| Quartz | — | 12 | — | 12 |
| Nitre | — | — | 4 | 4 |
| Lead got | 29.0 | 26.0 | 13.0 | 13.0 |
| Galena | 10 | 10 | 10 | 10 | 15 | 20 |
| Quartz. | — | 15 | — | 15 | 5 | — |
| Nitre | — | — | 3 | 3 | 3.5 | 7 |
| Lead got | 17.0 | 19.0 | 8.0 | 8.0 | 18.5 | 18.5 |
A similar set of experiments, with 80 grams of litharge instead of 80 grams of red lead, gave:—
| Pyrites | 4 | 4 | 4 | 4 | 7 | 10 |
| Quartz | — | 15 | — | 15 | 13 | 10 |
| Nitre | — | — | 5 | 5 | 12.5 | 20 |
| Lead got | 46.5 | 40.5 | 25.5 | 24.5 | 27.0 | 26.5 |
| Copper Pyrites | 5 | 5 | 5 | 5 | ||
| Quartz | — | 15 | — | 15 | ||
| Nitre | — | — | 5 | 5 | ||
| Lead got | 44.5 | 32.5 | 23.0 | 25.0 | ||
| Blende | 5 | 5 | 5 | 5 | 10 | |
| Quartz | — | 15 | — | 15 | 10 | |
| Nitre | — | — | 5 | 5 | 15 | |
| Lead got | 41.5 | 38.5 | 21.5 | 22.5 | 21.6 | |
| Antimonite | 5 | 5 | 5 | 5 | 10 | |
| Quartz | — | 15 | — | 15 | 10 | |
| Nitre | — | — | 5 | 5 | 10 | |
| Lead got | 31.0 | 32.5 | 11.5 | 12.5 | 18.7 | |
| Galena | 10 | 10 | 10 | 10 | 15 | 20 |
| Quartz | — | 15 | — | 15 | 5 | — |
| Nitre | — | — | 5 | 5 | 7.5 | 11 |
| Lead got | 33.5 | 33.5 | 13.0 | 14.0 | 19.5 | 22.7 |
The variation in some of these experiments, in which we might have expected similar results, is due to the fact that the sulphur, and in some cases the metals, are capable of two degrees of oxidation. For example: theoretically 1 gram of iron pyrites (FeS2) would yield 8.6 grams of lead if the sulphur were oxidised to sulphurous oxide (SO2), and the iron to ferrous oxide (FeO); whilst if the sulphur were oxidised to sulphate (SO3), and the iron to ferric oxide, 12.9 grams of lead will be thrown down. Similarly the yield with copper pyrites would be 7.5 or 11.6; with blende, 6.4 or 8.5; with antimonite, 5.5 or 8; and with galena, 2.6 or 3.4. As regards the metals, the lower oxide will always be formed if the assay is carried out properly (fused under a cover, and with a sufficiency of reducing agent). But the proportion of sulphur oxidised completely will vary with the conditions of the assay. With a slag containing much soda the tendency will be to form sulphate, and, in consequence, a big reduction of lead; whilst with an acid slag containing much quartz the tendency will be for the sulphur to go off as sulphurous oxide (SO2). In a fusion with litharge alone all the sulphur will be liberated as the lower oxide, whilst with much soda it will be wholly converted into sulphate. For example: 3 grams of an ore containing a good deal of pyrites and a little galena, gave, when fused with litharge, 16.5 grams of lead. A similar charge, containing in addition 20.0 grams of soda, gave 22.5 grams of lead.
It will be noted from the experiments that 1 gram of nitre kept up on the average 4 grams of lead; the range being from 3.2 with acid slags to 5.3 with very basic ones. These facts serve to explain some apparently irregular results got in practice.
The process is as follows:—The cupels, which should have been made some time before and stored in a dry place, are first cleaned by gentle rubbing with the finger and blowing off the loose dust; and then placed in a hot muffle and heated to redness for from 5 to 10 minutes before the alloy to be cupelled is placed on them. The reasons for this are sufficiently obvious: the sudden evolution of much steam will blow a cupel to pieces; and, if the whole of the water has not been removed before the cupel is filled with molten lead, the escaping steam will bubble through, and scatter about particles of the metal. If some particles of unburnt carbon remain in the bone ash, a similar result will be produced by the escape of bubbles of carbonic acid as soon as the fused litharge comes in contact with them. The cupels having been prepared are arranged in a definite order in the muffle, and the assay buttons[Pg 99] are arranged in a corresponding order on some suitable tray (cupel tray, fig. 41); the heat of the muffle being at bright redness. Then with the help of the tongs (fig. 42) the assay buttons should be placed each in its proper cupel; a note having been previously made of the position it is to occupy, and the door of the muffle closed.
This part of the work should be done promptly, so as not to unduly cool the muffle: the start requires a fairly high temperature, and is a critical part of the process. A black crust forms at once on the surface of the lead; but this ought soon to fuse and flow in greasy drops from off the face of the metal, so as to leave the latter fluid with a well-defined outline, and much brighter than the cupel. If this clearing does not take place, the buttons are said to be frozen; in which case the temperature must be raised, some pieces of charcoal put in the muffle, and the door closed. If they still do not clear, the heat must have been much too low, and it is best to reject them and repeat the assays.
When the buttons have cleared it is well to check the draught of the furnace, and to partly open the door of the muffle, so as to work at as low a temperature as is compatible with the continuation of the process.[11] Too low a temperature is indicated by the freezing of the buttons and the consequent spoiling of the assays. Experience soon enables one to judge when the heat is getting too low. A commoner error is to have the heat too high: it should be remembered that that which was high enough to clear the buttons at starting is more than sufficient to keep the process going. At the finish a higher temperature is again required: therefore the door of the muffle should be closed and the furnace urged. The finish is easily recognised. The drops of litharge which in the earlier stages flow steadily from the surface of the alloy, thin off later to a luminous film. At the end this film appears in commotion, then presents a brilliant play of colours, and, with a sudden extinction, the operation is finished. The metal again glows for an instant whilst becoming solid.
If the button is a small one the cupel is withdrawn at once and placed on that square of the cupel tray which corresponds to[Pg 100] the position it occupied in the muffle. If, however, it is fairly large precautions must be taken to prevent spirting.
Molten silver dissolves oxygen from the air and gives it off on solidifying; the escape of the gas on sudden cooling is violent and, by throwing off particles of the metal, may cause loss. This is called "vegetation" or "spirting." The silver is apparently solid when spirting takes place; the crust breaks suddenly and some of the metal is forced out. The evil is best guarded against by slow cooling and avoiding draughts. With large buttons of silver precautions should never be omitted. One plan is to allow the cupels to cool in the muffle itself, the mouth being closed with hot charcoal. Another is to cover the cupel with another cupel previously heated to redness; in this case the silver cools between two hot cupels, and, of course, cools slowly. A third plan is to withdraw the cupel to the door of the muffle, holding it until it begins to get solid and then immediately to put it back into the hotter part of the muffle.
Silver remains after cupellation in flattened elliptical buttons, adhering but only slightly to the cupel. Its upper surface should show faint markings as if it were crystalline. The presence of platinum renders it still more crystalline, but removes the characteristic lustre and renders the metal dull and grey. Copper, if not completely removed, has a very marked effect on the appearance of the button: the metal is spread out, damping, as it were, and firmly adhering to the cupel, which latter in the neighbourhood of the metal is almost black with oxide of copper. Sometimes the silver button is globular, or even more sharply rounded on its under than on its upper surface; it is said that this is due to the presence of lead. Gold may be present even to the extent of 50 per cent. without showing any yellow colour.
The appearance of the cupel affords some useful information. The presence of cracks evidently due to shrinkage indicates a badly made cupel. If, however, they are accompanied by a peculiar unfolding of the cupel, the margin losing its distinctness, it is because of the presence of antimony. When lead is the only easily oxidisable metal present, the stained portion of cupel is yellow when cold. A greenish tint may be due to small quantities of copper or, perhaps, nickel, cobalt, or platinum. Larger quantities of copper give a greenish grey or almost black colour. A dark green and corroded cupel may be due to iron. Rings of pale-coloured scoria may be due to tin, zinc, antimony, or arsenic. When the cupel shows signs of the presence of these metals in objectionable quantity, it is well to repeat the assay and scorify so as to remove them before cupellation.
The button should be detached from the cold cupel by seizing[Pg 101] with a pair of pliers: the under surface should be distorted by squeezing or hammering the button so as to loosen the adhering bone ash. The cleaning is easily completed by rubbing with a clean hard brush. After cleaning the buttons are best put on a tray of marked watch-glasses, and then taken to the balance and weighed. The weight of silver got needs a small correction; (1) by deducting for the amount of silver introduced by the lead or oxide of lead used in the assay;[12] and (2) by adding for the cupellation loss.
Loss in Cupellation.—During the whole process of cupelling a silver lead alloy a more or less abundant fume may be observed rising from the cupel. This furnishes an evident loss of lead and a possible loss of silver; for although silver at the temperature of cupellation gives off no appreciable vapour, it is known that such fume formed on a large scale contains silver. It is, however, difficult to believe that the small amount of lead vapourised carries with it a weighable amount of silver. That it does not do so in the ordinary way of working is shown by the fact that a button of silver equal in weight to the silver lost in cupelling may be got by smelting the cupel and cupelling the resulting button of lead. The loss of silver by volatilisation is altogether inconsiderable, unless the temperature at which the operation is performed is much too high.
Another possible source of loss is the infiltration of small particles of alloy into the cupel. The cupel is necessarily porous, and particles of metal may perhaps drain into it, more especially if the bone ash is not in fine powder; but if this is the main source of loss it is hard to see why, in cupelling equal weights of silver and gold, the loss is not equal in each case. It is not easy to believe that the mere filtration of the fused alloy will effect such a change in the proportion of the metals as that which actually occurs. For example: a cupel on which an alloy consisting of 0.80 gram of silver, 0.47 gram of gold, and 25 grams of lead had been cupelled, was found to contain 7-1/2 milligrams of silver, and rather less than half a milligram of gold. Assuming, for the sake of argument, that the gold present had filtered into the cupel in the form of small drops of alloy, it would have been accompanied by less than a milligram of silver, and the presence of the extra 6 or 7 milligrams of silver must have been due to a different cause. There can, thus, be little doubt that the cause of the greater part of the "cupellation loss" is a chemical one and cannot be counteracted by a mechanical contrivance.[13] In cupellation,[Pg 102] then, there is a loss, apart from imperfect working, inherent in the process itself; and as the amount of this loss varies under different conditions, it is necessary to study it somewhat in detail.
The following experiments are taken without selection from the work of one student. Three experiments were made for each determination, and the mean result is given. By "range" is meant the difference between the highest and lowest result and the percentage loss is calculated on the silver present. The silver added in the lead used has been deducted.
Effect of Varying Lead.—In each experiment 0.4 gram of silver was taken and cupelled with the lead. The silver loss and "range" are expressed in milligrams.
| Lead Used. | Silver Lost. | Range. | Percentage Loss. |
| Grams. | |||
| 10 | 6.5 | 1.0 | 1.62 |
| 20 | 7.0 | 1.0 | 1.75 |
| 40 | 12.0 | 1.5 | 3.00 |
| 60 | 12.7 | 0.5 | 3.17 |
The loss increases with the lead used.
Effect of Varying Temperature.—0.4 gram of silver was cupelled with 20 grams of lead.
| Temperature. | Silver Lost. | Range. | Percentage Loss. |
| Bright red | 7.0 | 1.0 | 1.75 |
| Clear yellow | 17.3 | 1.7 | 4.32 |
The difference in temperature in these experiments was much greater than would occur even with careless work.
Effect of Varying Silver.—20 grams of lead were used in each cupellation.
| Silver Taken. | Silver Lost. | Range. | Percentage Loss. |
| Milligrams. | |||
| 12.5 | 0.7 | 0.2 | 5.6 |
| 25.0 | 1.4 | 0.1 | 5.6 |
| 50.0 | 1.6 | 0.4 | 3.2 |
| 100.0 | 2.9 | 0.3 | 2.9 |
| 200.0 | 5.6 | 0.5 | 2.8 |
| 400.0 | 7.0 | 1.0 | 1.7 |
| 800.0 | 9.7 | 1.0 | 1.2 |
It will be seen that, although the quantity of silver lost increases[Pg 103] with the silver present, the percentage loss is greater on the smaller buttons.
The following results are often quoted:—Cupelling 1 grain of silver with 10 grains of lead, the loss was 1.22 per cent.; 10 grains of silver with 100 grains of lead, loss 1.13 per cent.; 25 grains of silver cupelled with 250 grains of lead, lost 1.07 per cent. The proportion of silver to lead was the same in the three experiments, and the largest button gave the best result. Evidently, if the quantities of lead had been the same in the three experiments (say, 250 grains in each case), the loss on the smaller quantities of silver would appear worse in the comparison.
In judging these results, it must be borne in mind that it is difficult to regulate the temperature, &c., in consecutive experiments so as to get exactly similar results, so that the range in consecutive cupellations is greater than that in a batch cupelled side by side.
Effect of Copper and Antimony.—0.1 gram of silver was cupelled with 20 grams of lead, and to one batch 0.5 gram of antimony, and to another 0.5 gram of copper was added.
| Silver Lost. | Range. | Loss in Percentage. | |
| Without addition | 2.9 | 0.3 | 2.9 |
| With antimony | 3.2 | 0.2 | 3.2 |
| With copper | 4.9 | 1.7 | 4.9 |
Perhaps the antimony has so small an effect because it is eliminated in the earlier part of the process, while the silver is still alloyed with, and protected by, a large proportion of lead; whilst the copper on the other hand makes its fiercest attack towards the close, when the silver is least capable of resisting it. The ill effects of copper are most strongly felt when the quantity of lead present is not sufficient to remove it: the coppery button of silver got under these conditions is very considerably less than the weight of silver originally taken.
Although the above is a fair statement of the loss attending average work, it will not do in very important and exact work to place too much reliance on the figures given, or, indeed, on any other set of figures, with the object of correcting the result of an assay. Each man must rely on his own work.
It is easy to determine what allowance must be made for the loss in cupellation by cupelling side by side with the assay piece an alloy of similar and known composition. For, if the two pieces are very nearly alike, we may justly conclude that the loss on each will be the same; and if, further, we take the average of three or four such determinations we shall get results accurate within 0.1 per cent. The method of getting such results may be best explained[Pg 104] by one or two illustrations. This method of working is termed "assaying by checks."
Suppose we have an alloy of silver and lead in unknown proportions and that by cupelling two lots of 10 grams each there is got from I. 0.1226 gram of silver, and from II. 0.1229 gram. We should know from general experience that the actual quantity of silver present was from 2 to 4 milligrams more than this. To determine more exactly what the loss is, the following plan is recommended:—The two silver buttons are wrapped up each in 10 grams of lead, and cupelled side by side with two other lots of 10 grams of the original alloy. If now the two buttons I. and II. weigh 0.1202 and 0.1203, they will have suffered in this second cupellation an average loss of 2.5 milligrams. Suppose the two fresh lots of alloy gave 0.1233 and 0.1235 of silver, the average loss on these would also be 2.5 milligrams. Add this loss to each result, and take the mean; which is in this case 0.1259.
If copper is present in the alloy as well as silver, it is necessary to add about the same quantity of copper to the checks as is supposed, or known, to be present in the assays. If the substance to be assayed is an alloy of silver and copper, first cupel 0.5 gram of it, with, say, 10 grams of lead, and weigh the resulting button of silver, in order to get an approximate knowledge of its composition. Suppose the button weighs 0.3935 gram. We know that this is below the truth: for the sake of round numbers take it as 0.4, and assume that the rest of the alloy (0.1 gram) was copper. Two check pieces are then weighed out, each containing 0.4 gram silver and 0.1 gram of copper wrapped in 5 grams of lead. Of course the silver must be pure. And there is also weighed out two (or better, four) assay pieces each containing half a gram of the alloy wrapped in 5 grams of lead. The whole lot are then cupelled as nearly as possible under the same conditions. With four assay pieces, the cupels should be placed close together in two rows of three across the muffle; the two check pieces are put in the middle cupels. Suppose the buttons of silver got weighed as follows:—
| Check pieces | I. | 0.3940 | II. | 0.3945 |
| Assay pieces | I. | 0.3905 | II. | 0.3912 |
| III. | 0.3910 | IV. | 0.3909 |
The average loss on the two check pieces is 5.7 milligrams, and the average result of the four assay pieces is 0.3909. Add the average loss to the average result, and there is got the corrected result, 0.3966. And if 0.5 gram of alloy contain 0.3966 of silver, 1000 will contain 793.2 of silver, and this is the degree of fineness.
A correction for the loss in cupellation is always made in this[Pg 105] way when rich alloys are being assayed; and in the case of rich ores it may be done after the manner of the first of the above illustrations. There is another method of working which relies more on experiment. This is to smelt the cupel as described further on (p. 114), and to again cupel the resulting button of lead. The button of silver got in this second cupellation is added to that first obtained. It will sometimes, but not often, happen that the two buttons together will slightly exceed in weight the silver which was actually present. This is because of the retention in the buttons of a small quantity of lead. It has been stated that the proportion of lead thus retained may be as much as 1% of the silver present; this, however, can only be under exceptional conditions. A determination of the actual silver in the buttons got in the series of cupellations quoted on pages 102, 103, gave an average percentage of 99.85, so that even with the larger buttons the effect of the retained lead would be only to increase the weight by about 1 milligram. In the method of working with checks, the retained lead has no disturbing influence.
The proportion of lead required for the cupellation of any particular alloy requires consideration. With too much lead the time occupied in the process is increased, and so is the loss of silver; on the other hand, too little lead is of greater disadvantage than too much. From 8 to 16 parts of lead are required for each part of silver alloy, or, if gold is present, about twice as much as this must be used. For the cupellation of 1 gram of a silver copper alloy containing different percentages of copper, the following quantities of lead should be used:—
| Percentage of Copper in Alloy. | Lead Required. | |
| 5 | 6 | grams |
| 10 | 8 | " |
| 20 | 10 | " |
| 30 | 12 | " |
| 40 | 14 | " |
| 50-100 | 16-18 | " |
The alloy, in not too large pieces, is wrapped in the required weight of lead foil and charged into the cupel at once; or the lead may be put in first, and, when the cupellation has fairly started, the alloy may be added wrapped in tissue paper; or a portion of the lead may be first started and the alloy wrapped in the remaining lead and subsequently added. The cupellation of large quantities of alloy or of alloys which contain tin, antimony, iron, or any substance which produces a scoria, or corrodes the cupel, must be preceded by a scorification. The advantages of this are that the slag is poorer in precious metal[Pg 106] than that found on a cupel and is more easily collected and cleaned; that larger quantities of metal can be treated, and that, even if the substance is in part infusible, or produces at the start a clinkery mass or scoria, the oxide of lead gradually accumulates, fluxes the solid matters, and produces a good final result; but if the oxide of lead by itself is not sufficient for the purpose, borax or some other flux can be easily added.
If the button of silver got is very small its weight may be estimated from its size; but it must be remembered that the weight varies as the cube of the diameter. If one button has twice the diameter of another it is eight times as heavy and so on. Scales specially constructed for measuring silver and gold buttons may be purchased; but it is much better to make the measurement with the help of a microscope provided with an eyepiece micrometer.
If the length of the long diameter of a silver button be taken the following table will give the corresponding weight in milligrams:—
| Diameter. | Weight. | Diameter. | Weight. |
| 0.04 inch | 3.6 | 0.015 inch | 0.19 |
| 0.035 " | 2.4 | 0.014 " | 0.15 |
| 0.03 " | 1.5 | 0.013 " | 0.12 |
| 0.025 " | 0.9 | 0.012 " | 0.097 |
| 0.02 " | 0.45 | 0.011 " | 0.075 |
| 0.019 " | 0.4 | 0.010 " | 0.056 |
| 0.018 " | 0.33 | 0.008 " | 0.028 |
| 0.017 " | 0.27 | 0.006 " | 0.012 |
| 0.016 " | 0.23 | 0.004 " | 0.004 |
The weight of a corresponding button of gold is got by multiplying by 2.25. These figures are based on those given by Plattner, and apply only to buttons of such shape as those left after cupellation. A sphere of silver 0.01 inch in diameter would weigh 0.09 milligram, and a similar sphere of gold weighs 0.167 milligram.
It is safer, however, to compare with a micrometer the diameter of the button whose weight has to be determined with that of a standard button of nearly equal size whose weight is known. The weights of the two buttons are proportional to the cubes of their diameters. This plan of working is described more fully in Appendix B., page 440.
Calculation of the Results.—After deducting for the silver added, and correcting for the cupellation loss, the calculation is made in the usual way; reporting as so many parts per thousand in the case of rich alloys and as so many ounces and[Pg 107] pennyweights, or better as ounces and decimals of an ounce, in the case of poor alloys and ores.
In this last case, however, it is less fatiguing to refer to a set of tables which give, either directly or by means of simple addition, the produce corresponding to any weight obtained from certain given weights of the substance. The following table gives the produce in ounces and decimals of an ounce per ton of 2240 pounds:—
| Weight of Metal got. | Weight of Ore taken. | ||||
| 3 grams. | 5 grams. | 20 grams. | 50 grams. | 100 grams. | |
| 0.0001 | 1.09 | 0.65 | 0.16 | 0.06 | 0.03 |
| 0.0002 | 2.18 | 1.31 | 0.33 | 0.13 | 0.06 |
| 0.0003 | 3.27 | 1.96 | 0.49 | 0.20 | 0.10 |
| 0.0004 | 4.36 | 2.61 | 0.65 | 0.26 | 0.13 |
| 0.0005 | 5.44 | 3.27 | 0.82 | 0.33 | 0.16 |
| 0.0006 | 6.53 | 3.92 | 0.98 | 0.39 | 0.19 |
| 0.0007 | 7.62 | 4.57 | 1.14 | 0.46 | 0.23 |
| 0.0008 | 8.71 | 5.23 | 1.31 | 0.52 | 0.26 |
| 0.0009 | 9.80 | 5.88 | 1.47 | 0.59 | 0.29 |
| 0.001 | 10.89 | 6.53 | 1.63 | 0.65 | 0.33 |
| 0.002 | 21.78 | 13.07 | 3.27 | 1.31 | 0.65 |
| 0.003 | 32.67 | 19.60 | 4.90 | 1.96 | 0.98 |
| 0.004 | 43.56 | 26.13 | 6.53 | 2.61 | 1.31 |
| 0.005 | 54.44 | 32.67 | 8.17 | 3.27 | 1.63 |
| 0.006 | 65.33 | 39.20 | 9.80 | 3.92 | 1.96 |
| 0.007 | 76.22 | 45.73 | 11.43 | 4.57 | 2.29 |
| 0.008 | 87.11 | 52.27 | 13.07 | 5.23 | 2.61 |
| 0.009 | 98.00 | 58.80 | 14.70 | 5.88 | 2.94 |
| 0.01 | 108.89 | 65.33 | 16.33 | 6.53 | 3.27 |
| 0.02 | 217.78 | 130.67 | 32.67 | 13.07 | 6.53 |
| 0.03 | 326.67 | 196.00 | 49.00 | 19.60 | 9.80 |
| 0.04 | 435.56 | 261.33 | 65.33 | 26.13 | 13.07 |
| 0.05 | 544.44 | 326.67 | 81.67 | 32.67 | 16.33 |
| 0.06 | 653.33 | 392.00 | 98.00 | 39.20 | 19.60 |
| 0.07 | 762.22 | 457.33 | 114.33 | 45.73 | 22.87 |
| 0.08 | 871.11 | 522.67 | 130.67 | 52.27 | 26.13 |
| 0.09 | 980.00 | 588.00 | 147.00 | 58.80 | 29.40 |
| 0.1 | 1088.89 | 653.33 | 163.33 | 65.33 | 32.67 |
| 0.2 | 2177.78 | 1306.67 | 326.67 | 130.67 | 65.33 |
| 0.3 | 3266.67 | 1960.00 | 490.00 | 196.00 | 98.00 |
| 0.4 | 4355.56 | 2613.33 | 653.33 | 261.33 | 130.67 |
| 0.5 | 5444.44 | 3266.67 | 816.67 | 326.67 | 163.33 |
| 0.6 | 6533.33 | 3920.00 | 980.00 | 392.00 | 196.00 |
| 0.7 | 7622.22 | 4573.33 | 1143.33 | 457.33 | 228.67 |
| 0.8 | 8711.11 | 5226.67 | 1306.67 | 522.67 | 261.33 |
| 0.9 | 9800.00 | 5880.00 | 1470.00 | 588.00 | 294.00 |
| 1.0 | 10888.89 | 6533.33 | 1633.33 | 653.33 | 326.67 |
When, as in this table, the fraction of an ounce is expressed by two places of decimals, it may be reduced to pennyweights (dwts.)[Pg 108] by dividing by 5. For example, 0.40 of an ounce is 8 dwts. The fraction of a dwt. similarly expressed may be converted into grains with sufficient exactness by dividing by 4. Thus, 1.63 ozs. equal 1 oz. 12.60 dwts., or 1 oz. 12 dwts. 15 grains. In England it is usual to report in ounces and decimals of an ounce.
The way to use the table is best shown by an example. Suppose a button of silver weighing 0.0435 gram was obtained from 20 grams of ore. Look down the 20-gram column of the table, and select the values corresponding to each figure of the weight, thus:—
0.04 = 65.33 ozs. to the ton
0.003 = 4.90 "
0.0005 = 0.82 "
——————
0.0435 = 71.05 "
Add these together. The produce is 71.05 ozs., or 71 ozs. 1 dwt. to the ton.
Or, suppose an ore is known to contain 1.24 per cent. of silver. Look down the 100-gram column, select the values, and add them together as before.
1.0 = 326.67 ozs. per ton
0.2 = 65.33 "
0.04 = 13.07 "
——————
1.24 = 405.07 "
This gives 405 ozs. 1 dwt. 10 grains to the ton.
The calculation becomes more complicated when, as is frequently the case, the ore contains metallic particles. These show themselves by refusing to pass through the sieve when the ore is powdered. When they are present, a large portion, or if feasible the whole, of the sample is powdered and sifted. The weights of the sifted portion and of the "metallics," or prills, are taken; the sum of these weights gives that of the whole of the sample taken. It is very important that nothing be lost during the operation of powdering.
Each portion has to be assayed separately. It is usual to assay a portion of the sifted sample, say, 20 or 50 grams, and to add to the produce of this its share of the "metallics." This way of calculating, which is more convenient than correct, is illustrated by the following example:—
| Weight of | whole sample | 400 | grams |
| Made up of | sifted portions | 399 | " |
| " | "Metallics" | 1 | " |
| ———— | |||
| 400 | " |
Twenty grams of the sifted portion, when assayed, gave 0.1050 gram of silver. The whole of the "metallics" scorified and cupelled gave 0.842 gram of silver. Since the 20 grams assayed was 1-20th of the whole, 1-20th part of the 0.842 gram of silver (from the metallics) must be added to its produce. We thus get 0.1471 gram (0.1050 + 0.0421).
Referring to the 20 gram column, we get—
0.1 = 163.33
0.04 = 65.33
0.007 = 11.43
0.0001 = 0.16
—————————
0.1471 = 240.25 ounces per ton.
A more legitimate method of calculation is as follows:—Calculate separately the produce of each fraction as if they were from different ores. Multiply each produce (best stated in per cents.) by the weight of the corresponding fraction. Add together the products, and divide by the weight of the whole sample. Taking the same example for illustration, we have:—
Metallics.—Weight 1 gram.
1 gram of it yielded 0.842 grams of silver.
∴ Produce = 84.2 per cent.
Produce multiplied by the weight is still 84.2.
Sifted Portion.—Weight 399 grams.
20 grams of it yielded 0.105 gram of silver.
∴ Produce = 0.525 per cent.
Produce multiplied by weight (0.525 × 399) is 209.475.
Add together; and divide by 400, the weight of the whole sample—
84.2
209.475
———-
400) 293.675 (0.7342
0.7342 is the total produce of the ore in per cents.
Referring to the 100-gram column in the table we find 239.84 ounces to the ton as the produce.
0.7 = 228.67
0.03 = 9.80
0.004 = 1.31
0.0002 = 0.06
———
239.84
Comparing this with the result calculated by the first method—viz., 240.26, we see that that was 0.38 oz., or between 7 and 8 dwts. too high.
With ores containing "metallics" it is of great importance to powder the whole of the selected sample without loss during the process; and of even greater importance to well mix the sifted portion, of which the last portions to come through the sieve are[Pg 110] apt to be more than ordinarily rich through the grinding down of some portions of the metallic prills.
Remarks on Cupellation.—Cupellation is at once the neatest and the most important of the dry methods of assaying. Its purpose is to remove easily oxidisable metals, such as lead and copper, from silver and gold, which are oxidisable with difficulty. Metals of the first class are often spoken of as base, and gold and silver as noble metals.
When lead is exposed to the action of air at a temperature a little above redness, it combines with the oxygen of the air to form litharge, an oxide of lead, which at the temperature of its formation is a liquid. Consequently, if the lead rests on a porous support, which allows the fused litharge to drain away as fast as it is formed, a fresh surface of the lead will be continually exposed to the action of the air, and the operation goes on until the whole of the lead has been removed. Silver or gold exposed to similar treatment does not oxidise, but retains its metallic condition; so that an alloy of lead and silver similarly treated would yield its lead as oxide, which would sink into the support, while the silver would remain as a button of metal.
The porous support, which is called a cupel(fig. 5), should absorb the slag (oxide of lead, etc.) just as a sponge absorbs water, but must be sufficiently fine-grained to be impervious to the molten metal. At first sight it appears difficult to filter, as it were, a fluid slag from a fluid metal; but an ordinary filter-paper damped with oil will allow oils to run through and yet retain the water; but damped with water it will allow water to run through and retain oils. Similarly, fused slags damp and filter through a cupel, but the molten metal not damping it withdraws itself into a button, which is retained. Although, of course, if the cupel is very coarse-grained the metal may sink into the hollows.
Copper, antimony, tin, and most other metals, form powdery oxides, which are not of themselves easily fusible, and it is necessary when these are present to add some solvent or flux to render the oxide sufficiently fluid. Fortunately, oxide of lead is sufficient for the purpose; hence, mixed oxides of copper and lead, provided the lead is present in proper proportion, form a fluid slag. In separating copper from silver or gold, advantage is taken of this fact; for, although we cannot cupel an alloy of copper and silver, it is easy to cupel an alloy of copper, silver and lead. If, however, the lead is not present in sufficient quantity, the whole of the copper will not be removed, and the button of silver, still retaining copper, will be found embedded in a coating of black oxide of copper. Copper oxidises less easily than lead does; and, consequently, the alloy which is being cupelled becomes relatively[Pg 111] richer in copper as the operation proceeds. It is on this account that the ill-effects of the copper make themselves felt at the close of the operation, and that the oxide of copper is found accumulated around the button of silver. Tin and antimony, on the other hand, are more easily oxidised; and the tendency of their oxides to thicken the slag makes itself felt at the commencement: if the button of alloy once frees itself from the ring or crust of unfused oxide first formed, the cupellation proceeds quietly, and leaves a clean button of silver in the centre. But in either case the cupellation is imperfect, and should be repeated with a larger proportion of lead. An unfused and, consequently, unabsorbed slag tends to retain small buttons of alloy or metal, and thus cause serious loss.
There is a principle underlying many of the phenomena of dry silver assaying which the student should endeavour to understand; and which serves to emphasise and explain some facts which without an explanation may present difficulties. If a button of melted lead be covered with a layer of slag rich in oxide of lead, and a second metal be added, this other metal distributes itself between the metal and slag in proportions which depend mainly upon the ease with which it is oxidised, and to a large extent upon the relative quantities of material present. Easily oxidisable metals such as zinc, iron, antimony and tin, will go mainly into the slag, and, if the proportion of the slag is large, very little will go into the metal. On the other hand, with metals oxidisable with difficulty, such as silver, gold, and platinum, the reverse holds true; nearly the whole of the metals will go into the lead, and very little into the slag. If, however, the slag be very rich, say in antimony, the lead will contain antimony; and, on the other hand, if the lead be very rich in silver, the slag will contain silver in appreciable quantity. Copper, which is near lead in the facility with which it is oxidised, will serve for the purpose of a detailed example. The results of actual analyses of metal and slag formed in contact with each other are shown in the following table:—
Percentage Composition of the Metal.
| Lead. | Copper. |
| 6.8 | 93.2 |
| 20.0 | 80.0 |
| 28.0 | 72.0 |
| 32.0 | 68.0 |
| 85.0 | 15.0 |
Percentage Composition of the Slag.
| Lead. | Copper. |
| 71.4 | 21.4 |
| 78.0 | 17.0 |
| 80.0 | 12.5 |
| 86.0 | 6.7 |
| 90.0 | 3.6 |
It will be seen from this table that the slag is always much richer in lead and poorer in copper than the metal with which it[Pg 112] is in contact. The ratio of lead to copper in these five samples is:—
| In the Metal. | In the Slag. | |
| 1 : 14 | 1 : 0.3 | |
| 1 : 4 | 1 : 0.2 | |
| 1 : 2.5 | 1 : 0.16 | |
| 1 : 2 | 1 : 0.08 | |
| 1 : 0.16 | 1 : 0.04 |
Assuming these figures to be correct, the following statement is approximately true. On oxidising an alloy of 10 grams of copper and 10 grams of lead, and pouring off the slag when 3 grams of lead have gone into it, there will be a loss of (owing to the slag carrying it off) about 0.2 gram of copper. On repeating the operation, the next 3 grams of lead will carry with them about 0.5 gram of copper; and on again repeating, 3 grams of lead will remove 0.8 gram of copper. Finally, the last gram of lead will carry with it 0.3 gram of copper, and there will be left a button of copper weighing 8.3 grams. The slag will have carried off altogether 1.7 gram of copper, which is 17 per cent. of the metal originally present.
With the more perfect exposure to the air, and quicker removal of the slag, which results from heating on a cupel, the loss would be heavier. Karsten got by actual experiment on cupelling copper and lead in equal proportions, a loss of 21.25 per cent.
Going back to the example: if the slag were collected and fused with a suitable reducing agent so as to convert, say, half of it into metal, that half would contain nearly the whole of the copper (such a reduction is called "cleaning the slag"). On reoxidising this metal, another button of copper is formed which, added to the first, would reduce the loss from 17 per cent. to, say, 7 or 8 per cent. And it is conceivable that by a series of similar operations, almost the whole of the 10 grams of copper originally taken might be recovered. In practice the problem is (as far as the copper is concerned) not how to save, but how most easily to remove it; and since the removal of this metal is quicker from an alloy containing not too much lead, it is evident that two or three operations with small quantities of lead will be more effectual than a single treatment with a larger quantity. With those metals (tin, antimony, &c.) which pass quickly into the slag, the contrary is true; hence with these it is necessary to have enough lead present, so that the slag formed at the outset shall contain enough oxide of lead to make it fluid. As silver is so much less easily oxidised than copper, we should reasonably expect that the proportion of silver carried off in the oxide of lead would be considerably less than that of the copper indicated in the above[Pg 113] example. Indeed, there are one or two facts which tend to encourage the hope that the operation may be conducted without any loss. If a piece of pure silver foil is exposed on a cupel to air at the usual temperature of cupellation, it undergoes very little change; it does not even fuse; it loses nothing in weight, and does not oxidise. In fact, even if oxide of silver were formed under these conditions, it could not continue to exist, for it is decomposed into silver and oxygen at a temperature considerably below redness. On the other hand, oxide of silver is not reduced to metal by heat alone, when mixed with an excess of oxide of lead; while metallic silver is converted into oxide when heated with the higher oxides of lead, copper, and some other metals. That silver, and even gold (which is more difficult to oxidise than silver), may be carried off in the slag in this way, is in agreement with general experience. If 10 grams of silver are cupelled with 10 grams of lead, there will be a loss of about 50 milligrams of silver, which is in round numbers 1-30th of the corresponding copper loss; with 10 grams of gold and 10 grams of lead, the loss will be 4 or 5 milligrams, which is about 1-12th of the corresponding silver loss.
Determination of Silver in Assay Lead.—Scorify 50 grams of the lead with 0.5 gram of powdered quartz or glass at not too high a temperature. When the eye has "closed in," pour; reject the slag, and cupel the button of lead. Remove the cupel from the muffle immediately the operation is finished. Weigh, and make a prominent note of the result in the assay book, as so many milligrams of silver contained in 100 grams of lead.
Determination of Silver in Red Lead or Litharge.—Fuse 100 grams of the oxide with from 10 to 20 grams of borax; and in the case of litharge with 2 grams or with red lead 4 grams of flour. Cupel the lead, and weigh the button of silver. Note the result as in the last case.
Determination of Silver in Argentiferous Lead.—Be careful in taking the sample, since with rich silver lead alloys the error from bad sampling may amount to several parts per cent. Cupel two lots of 20 grams each, and weigh the buttons of silver. Add to these the estimated cupel loss, and calculate the result. Or wrap each button of silver in 20 grams of assay lead, and re-cupel side by side with two fresh lots of 20 grams each of the alloy. Calculate the loss incurred, and add on to the weight of the two fresh buttons got.
Determination of Silver in Bullion.—The remarks made under the last heading as to the importance of correct sampling apply with equal force here. Make a preliminary assay by cupelling 0.1 gram of the alloy with 1 gram of assay lead; calculate[Pg 114] the percentage composition. Refer to the table on page 105 to find what weight of lead is required for cupelling 1 gram of alloy.
Weigh out four lots of 1 gram each, and wrap them in the required quantity of lead. Make two check pieces by weighing up two lots of fine silver equal to that which you believe to be present in the assay pieces; add copper to make up the weight to 1 gram, and wrap in the same quantity of lead as was used for the assays.
Prepare six cupels and charge them in the annexed order (fig. 43), and cupel. Guard against spirting. Clean and weigh the buttons of silver. Add the mean loss on the two check pieces to the mean weight of the four assay pieces; this multiplied by 1000 will give the degree of fineness.
Determination of Silver in Copper.—The silver is best separated in the wet way before cupelling, but if the proportion is not too small, it can be found by cupellation. Weigh up 3 grams of the metal, wrap in 30 grams of sheet lead, and cupel; when the cupellation has proceeded for fifteen minutes, add 20 grams more lead, and continue till finished. Weigh the button of silver.
The cupellation loss will be five or six per cent. of the silver present. Determine it by powdering the saturated portion of the cupel and fusing in a large Cornish crucible with 30 grams each of soda and borax, 10 grams of fluor spar, and 1-1/2 gram of charcoal. Cupel the resulting button of lead, and add 10 grams more of lead towards the close of the operation. Deduct the weight of silver contained in the lead used from the weight of the two buttons, and calculate to ounces to the ton.
In an experiment in which 0.1975 gram of silver was present, the weight of the button from the first cupellation was 0.1867, and that of the button from the second, after correcting for the lead added, was 0.0110 gram.
Determination of Silver in Galena. By Pot Assay.—Mix 20 grams of the powdered ore with 30 grams of red lead, 20 grams of soda, and 5 grams of borax, as also with from 7 to 10 grams of nitre. Fuse and pour. Clean the slag if the ore is rich. Cupel the buttons of lead. Make the usual corrections and calculate in ounces to the ton.
By Scorification.—Take 10 grams of the ore, 30 grams of lead,[Pg 115] and 0.5 gram of borax. Scorify, clean the slag by adding anthracite after the "eye" has closed in: cupel the button of lead. Weigh the button of silver, make the necessary corrections, and calculate to ounces to the ton.
The determination may also be made by cupelling the button of lead got in the dry lead assay.
A sample of galena determined by the three methods gave the following results:—
| By | pot assay | 7.18 ozs. | per ton. |
| " | scorification | 7.02 | " |
| " | lead assay | 6.72 | " |
Determination of Silver in an Ore. By Pot Assay.—Take 20 grams of the powdered ore and mix with 30 grams of soda, 40 grams of red lead, and 5 grams of borax, as also with from 2 to 3 grams of flour. Fuse: pour. Clean the slag by fusing with 20 grams of red lead and two grams of flour. Cupel the buttons of lead; weigh; make the necessary corrections, and calculate to ounces to the ton.
By Scorification.—Take 5 grams of the powdered ore, 50 grams of lead, and 0.5 gram of "soda" or borax. Scorify. Clean the slag by fusing in a crucible as in the pot assay. Cupel, &c.
Examples.—By Pot Assay.—Ore taken 20 grams.
| Silver got | 0.2893 | gram | |
| Silver from slag | 0.0060 | " | |
| Silver lost in cupellation | 0.0100 | " | |
| ——— | |||
| 0.3053 | " | ||
| Deduct silver in red lead | 0.0017 | " | |
| ——— | |||
| Silver in ore | 0.3036 | " | = 495.9 ozs. per ton. |
By Scorification.—Ore taken, 3 grams.
| Silver got. | 0.0425 | gram | |
| Silver from slag | 0.0022 | " | |
| Silver lost in cupellation | 0.0020 | " | |
| ——— | |||
| 0.0467 | " | ||
| Deduct silver in lead | 0.0015 | " | |
| ——— | |||
| Silver in ore | 0.0452 | " | = 492.2 ozs. per ton. |
Determination of Silver in Silver Precipitate.—This substance contains, in addition to metallic silver and gold, sulphates of lead and lime; oxides of zinc, copper, and iron; and more or less organic matter. The sample as received is generally free from "water at 100° C."; and, since it rapidly absorbs water, care should be taken in weighing it.[Pg 116]
Since it contains combined water it is not suited for scorifying; therefore the determination of silver and gold (fine metal) is made by pot assay. Weigh up 5 grams of the precipitate, mix with 100 grams of litharge and 1 gram of charcoal. Melt in a crucible at a moderate heat and pour. Detach the slag, replace in the crucible, and, when fused, add a mixture of 20 grams of litharge and 1 gram of charcoal. When the fusion is again tranquil, pour; and cupel the two buttons of lead.
In a sample worked in this manner the mean of four determinations gave 0.6819 gram of "fine metal"; deducting 1 milligram for the silver contained in the oxide of lead, and adding 8 milligrams for the cupellation loss, there is got 0.6889 gram or 13.778 per cent. of silver (and gold) in the sample.
Determination of Silver in Burnt Ores. By Pot Assay.—Roasted cupriferous pyrites containing small quantities of gold and silver comes under this heading. The following mixture will give a fluid slag which is heavy and tough when cold:—
| Ore. | Borax. | Sand. | Litharge. | Charcoal. |
| 100 | 50 | 50 | 100 | 7 |
Mix; place in a large crucible; cover with salt; and melt down under cover. When fused drop in an iron rod for a few minutes, and about a couple of minutes after its withdrawal, pour the charge quickly into a large conical mould. The button of lead should weigh about 50 grams. Cupel and weigh the silver. The litharge may be replaced by red lead, in which case another gram of charcoal powder must be added.
In our experience the results obtained by this method are about 20 per cent. less than the actual content of the ore. The results of two assays, after deducting for the silver in the litharge used, were 3.9 and 4.1 milligrams; and a third assay, in which 5.4 milligrams of silver had been added, gave 9.2, which, after deducting the added silver, leaves 3.8 milligrams. The average of the three results is 3.9 milligrams from the 100 grams of ore.
Two lots of 100 grams of the same ore treated in the wet way gave 5.2 and 5.0 milligrams of silver. Burnt ores from Spanish pyrites carry about 0.005 per cent. of silver.
Silver is got into solution from its ores by attacking with nitric acid, but it is best, after dissolving, to cautiously add dilute hydrochloric acid, and to carefully avoid excess. If the quantity of silver is very small the solution is allowed to stand twenty-four hours, but, otherwise, it is warmed and filtered as soon as it clears.[Pg 117] Dry the residue and concentrate the silver in a button of lead by pot method or scorification, according to the amount of stony matter present. Cupel the lead, and the resulting button will be free from all metals, except perhaps gold. It may be weighed; or dissolved in nitric acid, and the silver determined gravimetrically in the diluted and filtered solution. It is better to weigh the metal and afterwards to determine the gold in it, estimating the silver by difference. Silver alloys are dissolved in dilute nitric acid (free from chlorides), diluted, and filtered. The solution is then ready for gravimetric determination.
Sulphuretted hydrogen precipitates silver (like copper), completely, even from fairly acid solutions.
Add dilute hydrochloric acid in small excess to the hot dilute solution, which must contain free nitric acid. Heat and stir until the solution clears. Decant through a small filter, and wash with hot water, acidulated at first with a little nitric acid if bismuth is suspected to be present. Dry quickly, transfer as much as possible of the precipitate to a watch-glass; burn and ignite the filter paper, treating the ash first with two drops of nitric acid and then with one of hydrochloric, and again dry. Add the rest of the silver chloride and heat slowly over a Bunsen burner until it begins to fuse. Cool and weigh.
The precipitate is silver chloride (AgCl) and contains 75.27 per cent. of silver. The moist precipitate is heavy and curdy; it is decomposed by direct sunlight, becoming violet under its influence. When heated it is yellowish; and, since it is volatile at a high temperature, it must not, in drying, be heated above its fusing point. The fused chloride can be removed from the crucible (to which it adheres strongly) by digesting with dilute acid and zinc.
For the determination of silver in nearly pure bullion the following process is used:—Weigh up 1.5054 gram of the alloy. With this amount of alloy each 2 milligrams of silver chloride formed is equivalent to 1 degree of fineness, so that the weight of the silver chloride obtained (stated in milligrams and divided by 2) will give the degree of fineness. Transfer to a bottle (known as "bottles for the Indian mint assay") and dissolve in 10 c.c. of dilute nitric acid, then make up with water to 200 c.c. and add 3 c.c. of dilute hydrochloric acid. Allow to stand a few minutes and then shake. Fill the bottle completely with water, allow to settle, and syphon off the clear liquid; pour on more water,[Pg 118] shake gently to break up the lumps, and again fill the bottle with water. Invert over the mouth of the bottle a porous Wedgwood crucible, somewhat similar to those used in gold parting. Take firm hold of the crucible and bottle, and invert promptly so that the silver chloride may be collected in the crucible. Allow to stand a little while for the precipitate to settle, and then carefully remove the crucible under water.[14] Drain off most of the water and break up the silver chloride with the help of a well-rounded glass rod. This greatly facilitates the subsequent drying. Dry first on the water bath and then on the iron plate. Remove the dried silver chloride, by inverting the crucible, and weigh it.
As an example, 3 determinations of silver in a coin carried out in this way gave:—
| (1) | 1.8500 | gram AgCl | = 925.0 | fineness. |
| (2) | 1.8498 | " | = 924.9 | " |
| (3) | 1.8502 | " | = 925.1 | " |
Determination of Silver in Burnt Ores.—Take 100 grams of the ore and place in a large beaker of 2-1/2 litres capacity, and cover with 375 c.c. of hydrochloric acid. Boil for half an hour until the oxides are dissolved and the residue looks like sand and pyrites; then add 20 c.c. of nitric acid, and boil till free from nitrous fumes. Dilute to 2 litres with water, and pass a current of sulphuretted hydrogen till the iron is reduced, the copper and silver precipitated, and the liquor smells of the gas. This takes about one hour and a half.
Filter off the precipitate (rejecting the solution) and wash with warm water. Dry and transfer to an evaporating dish, adding the ashes of the filter paper. Heat gently with a Bunsen burner until the sulphur burns, and then calcine until no more sulphurous oxide comes off. When cold add 30 c.c. of nitric acid, boil and dilute to 100 c.c. Add 1 c.c. of very dilute hydrochloric acid (1 to 100),[15] stir well, and allow to stand overnight. Decant on to a Swedish filter paper, dry and calcine.
Mix the ashes with 100 grams of litharge and 1 gram of charcoal, and fuse in a small crucible. Detach the button of lead and cupel. Weigh and make the usual corrections. As an example, 100 grams of ore treated in this way gave 5.8 milligrams of silver; deducting 0.8 for the silver added in the oxide of lead leaves 5 milligrams obtained from the ore. Another experiment on 100 grams of the same ore to which 5 milligrams of silver had[Pg 119] been added gave 11.0 milligrams. Deduct 5.8 for the silver added; this leaves 5.2 milligrams as the silver obtained from the ore. These give, as a mean result, 0.0051 per cent., or 1.66 ounce per ton.
Determination of Silver in Commercial Copper.—For the method of doing this, with an example and experiment, see under the heading of Examination of Commercial Copper.
There are two of these, one adapted for the determination of silver in alloys of approximately known composition, and the other of more general application. The first of these, generally known as "Gay-Lussac's" method is, as regards its working, perfect in principle; but it requires a practically constant quantity of silver, that is, one which varies by a few milligrams only in each determination. It is a confirmatory method rather than a determinative one. The other is known as "Volhard's," and resembles in principle and method an ordinary volumetric process.
Gay-Lussac's method is based on the precipitation of silver from a nitric acid solution by a solution of sodium chloride. The point at which the whole of the silver is precipitated being recognised by the standard solution ceasing to give a precipitate. The process depends for its success upon, (1) the ease which silver chloride separates out from the solution leaving it clear after shaking, and, (2), the cloudiness produced by the reaction of very small quantities of silver nitrate and sodium chloride. In working, a quantity of the sodium chloride solution equal to 1 gram of silver is added at once to the assay; and, when the solution has been rendered clear by shaking, the residual silver (which should not exceed a few milligrams) is estimated with the help of a weaker solution of sodium chloride. The success in working evidently depends upon the accuracy with which the first addition of the salt solution is made. On this account the standard solution is run in from a special pipette capable of delivering a practically invariable volume of solution. It is not so important that this shall deliver exactly 100 c.c. as that in two consecutive deliveries the volume shall not differ by more than 0.05 c.c. The dilute salt solution is one-tenth of the strength of that first run in, and 1 c.c. of it is equivalent to 1 milligram of silver. Ordinarily it is run in 1 c.c. at a time (and an ordinary burette may be used for this purpose), shaking between each addition until it ceases to give a precipitate. If many such additions have to be made the operation not only becomes tedious, but the solution[Pg 120] also ceases to clear after shaking, so that it becomes impossible to determine the finishing point.
If the assay contains less than one gram of silver the first addition of the dilute salt solution of course produces no precipitate. Five milligrams of silver in solution (5 c.c.) is then added, and the assay proceeded with in the usual way; 5 milligrams of silver being deducted from the amount found.
There is required for the assay a standard solution of sodium chloride, which is prepared by dissolving 5.4162 grams of the salt (made by neutralizing carbonate of soda with hydrochloric acid) in water and diluting to one litre. 100 c.c. of this is equivalent to 1 gram of silver.
The weaker solution of salt is made by diluting 100 c.c. of the stronger one to one litre. One c.c. of this will equal 1 milligram of silver, or 0.1 c.c. of the stronger solution.
A standard solution of silver equivalent to the dilute salt solution is made by dissolving 1 gram of fine silver in 10 c.c. of dilute nitric acid, and diluting with water to one litre.
The solution of salt is standardised as follows:—Weigh up 1.003 gram of fine silver and dissolve in 25 c.c. of dilute nitric acid in a bottle provided with a well-fitting flat-headed stopper. Heat on the water bath to assist solution, resting the bottle in an inclined position. When dissolved blow out the nitrous fumes with the help of a glass tube bent at right angles. Run in from a stoppered pipette (as shown in fig. 44) 100 c.c. of the standard salt solution, and shake vigorously until the solution clears. Fill an ordinary burette with the weaker standard salt solution, and run 1 c.c. into the assay bottle, letting it run down the side so that it forms a layer resting on the assay solution. If any silver remains in solution a cloudy layer will be formed at the junction where the two liquids meet. This is best observed against a black background If a cloudiness is seen, shake, to clear the liquid, and run in another c.c. of salt, and continue this until a cloudiness is no longer visible. Deduct 1.5 c.c. from the amount of the weaker sodium chloride solution run in. Divide the corrected reading by 10, and add to the 100 c.c. This will give the volume of strong salt solution equivalent to the silver taken.
If the first addition of the weaker salt solution causes no cloudiness add 5 c.c. of the silver solution from an ordinary pipette, shake, and then run in the weaker salt solution, working as before. These 5 milligrams of silver added must be allowed[Pg 121] for before calculating. As an example:—1.0100 gram of fine silver was taken for standardising a solution and 4 c.c. of the weaker salt solution were run in. Deducting 1.5 and dividing by 10 gives 0.25 c.c. to be added to the 100 c.c.
100.25 : 1.0100 :: 100 : x
x = 1.0075
which is the standard of the salt solution.
The method of working an assay may be gathered from the following example:—In the determination of silver in some buttons left after cupellation, it was assumed that these would contain 99.5 per cent. of silver. For the assay it was necessary to take a quantity that should contain a little more than 1.0075 grams of silver; then
99.5 : 100 :: 1.0075 : x
x = 1.0125
To ensure a slight excess, there was taken 1.0150 gram of the buttons, which was treated in exactly the same way as for the standardising. The quantity of the weaker salt solution required was 7 c.c.; deducting 1.5 c.c., and dividing by 10, gives 100.55 c.c. of strong salt solution, which is equivalent to 1.0130 gram of silver. This being obtained from 1.015 gram of alloy, is equal to 99.8 per cent., or 998.0 fine.
The Effect of Temperature.—The standardising and the assay must be done at the same time, since a difference of 5° C. makes a difference of 0.1 c.c. in measuring the 100 c.c. of strong solution of salt. It is always best to prepare a standard with each batch of assays.
SULPHOCYANATE METHOD.—Volhard's process is based upon the precipitation of silver in nitric acid solutions with potassium sulphocyanate, the finishing point being the development of a reddish-brown colour, produced by the action of the excess of sulphocyanate upon ferric sulphate. The white sulphocyanate settles readily, leaving the liquor clear; and a persistent brown coloration in the liquid indicates the finish. The assay must be carried out in the cold; and water free from chlorides[16] must be used.
The standard sulphocyanate of potassium solution is made by dissolving 4-1/2 or 5 grams of the salt (KCyS) in water, and diluting to 1 litre. 100 c.c. are about equivalent to 0.5 gram of silver.[Pg 122]
The standard silver nitrate solution is made by dissolving 5 grams of fine silver in 50 c.c. of dilute nitric acid, boiling off nitrous fumes, and diluting to 1 litre.
The indicator is a saturated solution of iron alum, or a solution of ferric sulphate of equivalent strength made by titrating acid ferrous sulphate with potassium permanganate. Use 2 c.c. for each assay.
The sulphocyanate solution is standardised by placing 50 c.c. of the silver nitrate solution in a flask with 20 c.c. of dilute nitric acid, diluting to 100 c.c. with water, and running in the sulphocyanate until the greater part of the silver is precipitated; then adding 2 c.c. of the ferric indicator, and continuing the titration until a reddish-brown colour is developed, and remains permanent after shaking continuously. The assay is similarly performed, the silver being used in the state of a nitric acid solution.
The effect of variations in the conditions of the assay may be seen from the following experiments, in which 20 c.c. of standard silver nitrate were used:—
Effect of Varying Temperature:—
| Temperature | 10° C. | 30° C. | 70° C. | 100° C. |
| Sulphocyanate reqd. | 19.6 c.c. | 19.3 c.c. | 19.0 c.c. | 18.6 c.c. |
Effect of Varying Nitric Acid:—Varying nitric acid has no effect, except that with a fairly acid solution the finishing point is somewhat sharper.
| Nitric acid added | 5 c.c. | 10 c.c. | 20 c.c. | 50 c.c. |
| Sulphocyanate reqd. | 19.6 c.c. | 19.5 c.c. | 19.6 c.c. | 19.6 c.c. |
Effect of Varying Bulk:—
| Bulk | 50 c.c. | 100 c.c. | 200 c.c. | 300 c.c. |
| Sulphocyanate reqd. | 19.5 c.c. | 19.6 c.c. | 19.6 c.c. | 19.7 c.c. |
Effect of Varying Ammonic Nitrate:—
| Ammonic nitrate | 0 gram | 1 gram | 5 grams | 10 grams |
| Sulphocyanate reqd. | 19.6 c.c. | 19.6 c.c. | 19.7 c.c. | 19.9 c.c. |
Effect of Varying Silver:—
| Silver added | 1 c.c. | 10 c.c. | 20 c.c. | 50 c.c. | 100 c.c. |
| Sulphocyanate reqd. | 1.0 c.c. | 9.70 c.c. | 19.6 c.c. | 49.4 c.c. | 99.0 c.c. |
This method is valuable for determining silver in salts, alloys, and solutions, where no more than an ordinary degree of accuracy is demanded. It is easy, and applicable under most of the usual conditions. Its greatest disadvantage is the brown coloration[Pg 123] produced by the sulphocyanate when the assay is nearly, but not quite, finished; and the slowness with which this is removed on shaking up with the precipitate. This is worse with large quantities of precipitate, and if about 1 gram of silver is present, it gives an indefiniteness to the finish which lowers the precision of the process to about 1 in 500; this is useless for the assays of bullion. One writer states that this inconvenience is due to portions of liquid being entangled in the precipitate, but it appears much more likely to be due to the action of the precipitate itself. In attempting to apply the process to the assay of bullion by working it on the principle of a Gay-Lussac assay, it was found that a very considerable excess of silver was required to complete the reaction. In these experiments 100 c.c. of "sulphocyanate" (very accurately measured) was run into the solution containing the weighed portion of bullion (fine silver) and, after shaking the solution, was filtered. In the filtrate the remaining silver, if there should be any, was determined by the ordinary titration, but with "sulphocyanate" of one-tenth the strength. This final titration was quite satisfactory. The amount of silver precipitated by the first 100 c.c., however, varied with the quantity of silver present as in the following series.[17]
| Silver present. | Silver precipitated. | ||
| 1.1342 | gram. | 1.1322 | gram. |
| 1.1375 | " | 1.1335 | " |
| 1.1405 | " | 1.1351 | " |
| 1.1484 | " | 1.1379 | " |
These, of course, preclude a method of the kind aimed at, and at the same time emphasise the importance of uniformity of work in the ordinary process. In the determination of chlorides in sea-water, Dittmar used a combined method: precipitating the bulk of the silver as chloride, and after filtering, determining the small excess of silver by sulphocyanate. This modification answers admirably when applied to the assay of bullion. In the ordinary Gay-Lussac method, the precipitation of the bulk of the silver by the 100 c.c. of salt solution leaves nothing to be desired, either as to ease in working or accuracy of result; the silver precipitate settles quickly, and leaves a clear liquor admirably fitted for the determination of the few milligrams of silver remaining in solution. But the method of determining this residual silver by adding successive small quantities of salt so long as they continue to give a precipitate is unsatisfactory, and,[Pg 124] judged on its own merits apart from the rest of the process, could hardly escape condemnation. It is clumsy in practice, for the continued adding of small portions of salt solution is laborious and becomes impossible with more than a few milligrams of silver in solution. The proposed modification is simple; having precipitated the silver with the 100 c.c. of salt solution, as described under Gay-Lussac's method (page 120), shake till the liquor clears, and filter into a flask, washing with a little distilled water. Add 2 c.c. of "ferric indicator" to the filtrate and titrate with a standard "sulphocyanate solution" made by diluting the ordinary standard solution to such an extent that 100 c.c. after diluting shall be equivalent to 0.1 gram of silver.[18] Calculate the weight of silver found by "sulphocyanate" and add it to the weight which 100 c.c. of the salt solution will precipitate.
An advantage of this modification is that an excess of 15 milligrams may be determined as easily and exactly as 5. In standardising the salt solution, then, weigh up, say 1.0150 gram of pure silver, dissolve and titrate. Suppose 13.5 c.c. of "sulphocyanate" required; then these are equivalent to .0135 gram of silver, (100 c.c. = .1); the silver precipitated by the salt is 1.0150-.0135—i.e., 1.0015 gram, which is the standard.
Application of the Method to Assays for Arsenic.—If silver nitrate be added to a neutral solution of an arsenate of one of the alkali metals, silver arsenate (Ag3AsO4), is thrown down as a dark-red precipitate. If, after adding excess of silver nitrate to insure a complete precipitation, the arsenate of silver be filtered off, the weight of the arsenic could be estimated from the weight of silver arsenate formed. But this may be done much more conveniently by dissolving the precipitate in nitric acid, and titrating with sulphocyanate; the silver found will be to the arsenic present as 324 (108×3) is to 75.
The mineral is best treated by the method given in the third paragraph on page 382; but the solution, after being acidified with nitric acid, should be made exactly neutral with ammonia. A small excess of silver nitrate should then be added, and since acid is liberated in the reaction, the liquor must again be neutralised.[19] The precipitate must then be filtered off, and washed with distilled water. Then dissolve it in the paper by slowly running over it 20 c.c. of dilute nitric acid. Wash the filter with distilled water, collecting with the filtrate in a small flask. Add 2 c.c. of "ferric indicator" and titrate.[Pg 125]
If the sulphocyanate solution be made up with 11 or 12 grams of the potassium salt to the litre, and be then standardised and diluted, so that for 100 c.c. it shall equal 1.08 gram of silver, (see p. 38), then it will also equal .25 gram of arsenic (As). Except for ores rich in arsenic, it will be better to work with a solution one half this strength. The standard as calculated from an experiment with pure silver should be checked by another using pure resublimed white arsenic, As2O3, which contains 75.75 % of the metal. The quantity of white arsenic taken, .1 or .2 gram, should contain about as much arsenic as will be present in the assays. It is converted into sodium arsenate by evaporating to a small bulk with nitric acid and neutralising with soda. The precipitation and titration of the silver arsenate should be exactly as in the assays.
The difficulty of the method is in the neutralising; which has to be very carefully done since silver arsenate is soluble in even faintly acid solutions; one drop of nitric acid in 100 c.c. of water is enough to produce an absolutely worthless result; and an excess of acid much less than this is still very prejudicial. The addition of a little sodium acetate to the solution after the final neutralising has a good effect.
Arsenic in Mispickel.—Weigh up .250 gram of the finely-powdered ore, and place in a Berlin crucible about 1-1/4 or 1-1/2 inch in diameter. Treat with 10 or 12 drops, one drop at a time, of strong nitric acid, warm very gently, but avoid much heating. Put on a thin layer of nitre, and rather more than half fill the crucible with a mixture of equal parts of soda and nitre. Heat quickly in the blow-pipe flame, and when the mass is fused and effervescing, withdraw and allow to cool. Boil out with water, filter and wash. Insert a piece of litmus paper and cautiously neutralise with nitric acid, using ammonia to neutralise any accidental excess of the acid. Add a gram or so of ammonium nitrate and silver nitrate in excess, neutralise again with ammonia and add two or three grams of sodium acetate. Filter off the precipitate, wash and titrate. In the fusion care should be taken to avoid much effervescence (an excess of the soda mitigates this) and the operation should be stopped as soon as the whole has entered into fusion.
There is, properly speaking, no colorimetric method, but the following, which is sometimes used, is based on similar principles.[Pg 126] It is useful for the determination of small quantities of silver in substances which yield clear solutions with nitric acid.
Dissolve a weighed quantity of the substance in nitric acid, and dilute to a definite bulk. Divide into two equal parts. To one, add a drop or two of dilute hydrochloric acid, stir and filter. To the other, add a similar amount of dilute acid, and then to the filtered portion run in from a burette standard silver nitrate (1 c.c. = 0.5 milligram silver) until the solutions are equally turbid. Calculate in the usual way.
Gold occurs in nature chiefly as metal. It always contains more or less silver, and, in alluvial sands, &c., may be associated with platinum and iridium.
Gold is insoluble in hydrochloric or nitric acid, but is dissolved by aqua regia or by solutions of iodine, bromine, or chlorine. It is taken up by mercury, forming an amalgam, from which the mercury may be driven off by heat.
When gold occurs in particles of any size, it is readily detected by its appearance, but when finely disseminated through a large quantity of rock, it is separated and detected by the amalgamation assay—described below—or by a process of washing somewhat similar to vanning, or by the following test:—Powder and, if necessary, roast 50 to 100 grams of the ore, put on it three or four crystals of iodine and enough alcohol to cover it; allow to stand for half an hour; a piece of filter paper moistened with the liquid and burnt leaves an ash with a distinctly purple tint if any gold is present. It is better, however, to filter off the solution, evaporate, and ignite. Then, either take up with mercury, and ignite the amalgam so as to get a speck of the metallic gold; or treat with a few drops of aqua regia, and test the solution with stannous chloride: a purple coloration indicates gold.
AMALGAMATION ASSAY.—This does not attempt to give the total produce of gold, but rather the quantity which can be extracted on a large scale; therefore it should imitate as closely as possible the process adopted in the mine or district for extracting the metal.
Take 2 lbs of the ore in powder and roast; make into a stiff paste with hot water and rub up for an hour or so with a little mercury. Wash off the sand carefully, and collect the amalgam. Drive off the mercury by heat, and weigh the residual gold. It is best to cupel it with lead before weighing.[Pg 127]
In an experiment on a lot of ore which contained 0.189 gram of gold, 0.179 gram was obtained by the above process, equal to about 94-1/2 per cent. recovered. With ores generally, the yield may be from 80 to 90 per cent. of the actual gold present.
The dry assay of gold ores resembles in its main particulars the dry assay for silver by the crucible method; and for much that is of importance in its discussion the student is referred to what is written under Silver on pp. 90-113.
Size of Assay Charges.—Gold ores rarely contain more than a few ounces, often only a few pennyweights of gold to the ton; consequently, the button of gold obtainable from such quantities of ore as may be conveniently worked by assaying methods is often so small as to require more than ordinary care in its manipulation. One milligram of gold forms a button of about the size of one of the full-stops on this page, and compared with a million similar particles of quartz (about four ounces), represents a produce of a quarter of an ounce to the ton: a proportion such as the assayer is frequently called on to determine. It is evident, therefore, that a charge of half an ounce or less of the ore, such as is usual with silver ores, would demand of the worker both skill and care in the handling of the minute quantity of gold to be obtained from it. Fortunately the work is simple and precise, so that in practised hands and with only a 5-gram charge the assay of a 5-dwt. ore is practicable; with so small a charge, however, the result is barely perceptible on a sensitive balance: the button of gold should be measured under a microscope. It follows, therefore, that larger charges of say 50, 100, or even 200 grams, have an advantage in that they lessen the strain on the worker's attention, and, except in the case of the poorest mineral, bring the button of gold within the scope of the balance. On the other hand, the inconvenience of the larger charges lies in the amount of fluxes and consequent size of the crucibles required to flux them.
Sampling.—A further consideration in favour of the larger charges is the matter of sampling. In preparing his ore, the student should ask himself what reasonable expectation he has that the portion he puts in the furnace will be of average richness. The larger charges are likely to be nearer than the smaller ones to the average of the parcel of ore from which they are taken. In explanation of this, let us suppose a large[Pg 128] heap of 5-dwt. ore, in sand of the coarseness of full-stops, and containing all its gold in particles of 1 milligram, as uniformly distributed as care and labour in the mixing can accomplish. Such a heap could not possibly occur in practice, but it will serve for purposes of illustration. Now, one ton of the sand, however taken, would contain appreciably the same quantity of gold as any other ton. For a ton would contain about 8000 particles of gold; and even if two separate tons differed by as much as 100 particles (which they are just likely to do), this would mean only a difference of 1 or 2 grains to the ton. On the other hand, two portions of 14 lbs., which should contain on the average 50 particles of gold, are likely enough to differ by 10 particles, and this, calculated on a ton, means a difference of 1 dwt. It is easy to see that something like this should be true; for on calculating the 14-lb. lot up to a ton, the deviation from the average, whatever it may be, is multiplied by 160; whereas, if the ton were made up by adding 14-lb. lot to 14-lb. lot, up to the full tale, then a large proportion of the errors (some being in excess and some in defect) would neutralise each other. An average which is practically true when dealing with thousands, and perhaps sufficiently exact with hundreds, would be merely misleading when applied to tens and units. Reasonable safety in sampling, then, is dependent largely on the number of particles of gold in the charge taken, and the risk of an abnormal result is less, the larger the charge taken.
By doubling the charge, however, we merely double the number of particles. Powdering finely is much more effective; for, since the weight of a particle varies as the cube of the diameter, halving the diameter of the particles increases their number eight-fold. If, now, we modify our illustration by assuming the particles to have only one-sixth the diameter of a full-stop (which would represent a powder of a fineness not unusual in ores prepared for assaying), we should multiply the number of particles by 200 (6 × 6 × 6 = 216). We should then reasonably expect a 14-lb. parcel of the powder to give as safe a sample as a ton of the sand would give; and portions of a size fit for crucible work, say 50 or 100 grams, would be as safe as 10 or 20-lb. samples of the coarser stuff. For example, 60 grams of such powder would contain, for a 5-dwt. ore, about 100 particles; and in the majority of cases the error due to sampling would be less than 10 or 12 grains to the ton, and would only occasionally exceed a pennyweight. With richer ores the actual deviation stated as so much to the ton of ore might be greater, but it would represent a smaller proportion, stated in percentage of the[Pg 129] gold actually present, and would ultimately fall within the limits of unavoidable error.
It will be seen that the size of the quartz particles has no direct bearing on the argument; and, in fact, the coarseness of the quartz only interferes by preventing the uniform mixing of the sand and by binding together several particles of gold; in this last case, particles so united must, of course, count as one larger particle. Now, there are some natural ores in which the gold particles are all very small; with these fine powdering and mixing yields a product from which a sample may be safely taken. Then, again, in "tailings," before or after treatment with cyanide, we have a similar material, inasmuch as the coarser gold has been removed by previous amalgamation. With these, it is not unusual to take the portion for assay without any further powdering, since they are poor in gold, and have already been stamped and passed through a sieve of say thirty holes to the inch (linear).
But there are other ores, in lump showing no visible gold, which contain the gold in all possible degrees of fineness, from say prills of a milligram or so down to a most impalpable powder. The treatment of these cannot be so simple and straightforward. Suppose a parcel of 1000 grams (say 2 lbs.) of such ore in fine powder, containing on an average 1 particle of 1 milligram (the presence or absence of which makes a difference of .6 dwt. on the ton), 10 others of about .5 milligram (each representing .3 dwt.), and 100 others, which are too coarse to pass through an 80 sieve, and having an average weight of .1 milligram (each .06 dwt.), and that the rest of the gold, equivalent altogether to 2 ounces to the ton, is so finely divided that a charge of 50 grams may be taken without any considerable risk of its interfering with the sampling. Then in a 50-gram charge there would be one chance in twenty of getting the milligram particle, in which case the result would be 12.35 dwts. too high; on the other hand, if it were not present the result would on this account be .65 dwt. too low. Of the ten .5-milligram particles, it is as likely as not that one will be present, and its presence or absence would cause an error of 3.3 dwts., more or less. Of the 100 particles of .1 milligram, there would probably be from 3 to 7, instead of 5, the proper number; this would mean a variation of 2.6 dwts. from the true proportion. So that the probable result would range about 5 dwts. more or less than the 2-1/2 ozs., which is the true produce, and there are possibilities of astounding results. It is true that the majority of the results would be well within these limits, and now and again the heart of the student would be gladdened by a beautiful concordance in[Pg 130] duplicate assays; nevertheless, there can be no reasonable expectation of a good assay, and to work in this way, on a 50-gram charge, would be to court failure. The coarse gold must ruin the assay.
The difficulty may be met by concentrating the whole of the coarse gold in a small fraction of the ore, by sifting and making a separate assay of this fraction. A portion of the ore, of about 1000 grams, is ground to a very fine powder and passed through an 80 sieve, re-grinding when necessary, until only 20 or 30 grams is left of the coarser powder. This is mixed with fluxes and carried through as a separate assay. The sifted portion is thoroughly mixed, and a portion of it, say 30 or 50 grams, taken for assay. The weights of the two portions must be known, and care must be taken that nothing is lost in the powdering. The method of calculating the mean result from the two assays is shown on page 109. In this way of working there is no advantage in continuing the grinding until the coarser fraction is reduced to a gram or so—rather the contrary; and rubbing on until all the gold is sent through the sieve is to be distinctly avoided. The student must bear in mind that what he is aiming at is the exclusion of all coarse gold from the portion of ore of which he is going to take only a fraction.
The question of the smaller sampling of gold ores has been dwelt on at considerable length, as befits its importance, in order that the student may be impressed with a sense of its true meaning. Sampling is not a mystery, nor does the art lie in any subtle manner of division. It is, of course, absolutely necessary that the stuff to be sampled shall be well mixed, and the fractions taken, so that each part of the little heap shall contribute its share to the sample. Moreover, it must be remembered that tossing about is a poor sort of mixing, and that everything tending to separate the large from the small, the light from the heavy, or the soft from the hard (as happens in sifting), must be avoided, or, if unavoidable, must be remedied by subsequent mixing.
With a well-taken sample, we may rely on a great majority of our results falling within normal limits of error; but nothing can be more certain than that, in a moderately large experience we shall get, now and again, deviations much more considerable. These erratic assays can only be met by the method of working duplicates, which call attention to the fault by discordant results. Such faulty assays should be repeated in duplicate, so that we may rest the decision on three out of four determinations.
The likelihood of two very faulty assays being concordant is[Pg 131] remote; but with very important work, as in selling parcels of ore, even this risk should be avoided, as concordance in these cases is demanded in the reports of two or more assayers. The following actual reports on a disputed assay will illustrate this: (a) 5 ozs. 1 dwt.; (b) 5 ozs. 10 dwts. 12 grains; (c) 5 ozs. 11 dwts.; (d) 5 ozs. 11 dwts. 12 grs. The mean result of several assays, unless there be some fault in the method, will be very fairly exact; and individual assays, with an uncertainty of 1 in 20, may, by repetition, have this reduced to 1 in 100 or less.
Assay Tons, etc.—Having decided on taking a larger or smaller portion, the exact quantity to be used will be either some round number of grams, such as 50 or 100, easily calculable into percentage; or it will be that known as the "Assay Ton" (see page 13) or some simple multiple or fraction of it, which is easily calculable into ounces. The reports, too, are at least as often made as ounces in the short ton of 2000 lbs., as on the more orthodox ton of 2240 lbs. Now the short ton is equal to 29,166.6 troy ounces; and the corresponding "assay ton" is got from it by replacing ounces by milligrams. The advantage of its use is that if one assay ton of ore has been taken, the number of milligrams of gold obtained is also the number of ounces of gold in a ton of the ore, and there is absolutely no calculation. Even if half an assay ton has been taken the only calculation needed is multiplying the milligrams by two. On the other hand with a charge of two assay tons the milligrams need halving. Where weights of this kind (i.e., assay tons) are not at hand they may be easily extemporised out of buttons of tin or some suitable metal, and it is better to do this than to array out the grams and its fractions at each weighing. The sets of "assay tons," however, are easily purchased. As stated on page 13, the assay ton for 2240 lbs. is 32.6667 grams; and for the short ton, 29.1667 grams. If, however, the round number of grams be used and the result brought by calculation to the produce on 100 grams, the conversion to ounces to the ton may be quickly effected by the help of the table on page 107. As this table only deals with the ton of 2240 lbs., it is supplemented here by a shortened one dealing only with the produce of 100 grams and stating the result in ounces troy to the short ton of 2000 lbs.
Estimation of Small Quantities of Gold.—By the Balance. In estimating minute quantities of gold there are one or two points, of importance to an assayer only in this assay, where they will often allow one to avoid the working of inconveniently large charges. One of these is known as "weighing by the method of[Pg 132]
TABLE FOR CALCULATING OUNCES TO THE SHORT TON FROM THE YIELD OF GOLD FROM 100 GRAMS OF ORE.
| Milligram. | Ounces to the Ton. | Milligram. | Ounces to the Ton. | Milligram. | Ounces to the Ton. |
| 0.01 | 0.003 | 0.4 | 0.117 | 7.0 | 2.042 |
| 0.02 | 0.006 | 0.5 | 0.145 | 8.0 | 2.333 |
| 0.03 | 0.009 | 0.6 | 0.175 | 9.0 | 2.625 |
| 0.04 | 0.012 | 0.7 | 0.204 | 10.0 | 2.916 |
| 0.05 | 0.014 | 0.8 | 0.233 | 20.0 | 5.833 |
| 0.06 | 0.017 | 0.9 | 0.262 | 30.0 | 8.750 |
| 0.07 | 0.020 | 1.0 | 0.292 | 40.0 | 11.666 |
| 0.08 | 0.023 | 2.0 | 0.583 | 50.0 | 14.583 |
| 0.09 | 0.026 | 3.0 | 0.875 | 60.0 | 17.500 |
| 0.10 | 0.029 | 4.0 | 1.167 | 70.0 | 20.416 |
| 0.20 | 0.058 | 5.0 | 1.458 | 80.0 | 23.333 |
| 0.30 | 0.087 | 6.0 | 1.750 | 90.0 | 26.250 |
vibrations." Suppose a balance at rest in perfect equilibrium, with the pointer exactly over the middle point of the scale. Let the scale be a series of points at equal distances along a horizontal line; then, if a small weight be placed on one pan, the pointer will deviate from its vertical position and come to rest opposite some definite part of the scale, which will depend upon the magnitude of the weight added. The law determining this position is a very simple one; the deviation as measured along the points of the scale varies directly as the weight added. For example, with an ordinarily sensitive balance, such as is used for general purposes, one milligram will move the pointer along, say, three divisions of the scale; then two milligrams will move it six divisions; half a milligram, one and a half divisions; and so on. Of course, with a more sensitive balance the deviations will be greater. Now the point at which the needle comes to rest is also the middle point about which it vibrates when swinging. For example, if the needle swings from the third to the seventh division on the right then [(7+3)/2] it will come to rest on the fifth. In working by this method the following conventions are useful: Always place the button to be weighed on the left pan of the balance, the weights on the right; count the divisions of the scale from the centre to right and left, marking the former + and the latter -; thus -5 is the fifth division to the left. Then the position of rest is half the algebraic sum of two readings. For example, let the readings be 7 to the right and 3 to the left, then (+7-3)/2 = +2. The mean division is the second division[Pg 133] to the right. If the student will place himself in front of a balance and repeat the following observations and replace the figures here given by his own, he will have no difficulty in grasping the method. First determine the bias of the balance; suppose the unloaded balance swings +1.25 and -1; the bias then is (1.25-1)/2 = +.125 or one-eighth of a division to the right. Now having put on the button to be weighed let the readings be +7.5 and +9.25, and (7.5+9.25)/2 = +8.375. Then the effect of the button has been to move the pointer from +.125 to +8.375, or 8.25 divisions to the right; we should, therefore, add the weight equivalent of 8.25 divisions to the weights, whatever they may be on the right hand pan of the balance; if the divisions were to the left (- divisions) we should subtract. The value of 1 division is easily determined. Suppose the button in the example were a 1 milligram weight, then we should have found that 1 milligram = 8.25 divisions ∴ 1 division = .121 milligram. This method of working adds very considerably to the power of a balance in distinguishing small quantities.
By the Microscope.—The use of the microscope also is a real advantage in estimating the weights of minute buttons of gold where there is no undue risk in sampling, and where an error of say 1 in 20 on the quantity of gold is tolerable. For ores with copper, lead, zinc, &c., as well as for tailings rather poor in gold, this leaves a wide field of usefulness. The method is described on page 440, but the description needs supplementing for those who are not accustomed to the use of a microscope. The eye-piece of a microscope (fig. 44a, A) unscrews at a, showing a diaphragm at b, which will serve as a support for an eye-piece micrometer. This last, B, is a scale engraved on glass, and may be purchased of any optical instrument maker, though it may be necessary to send the eye-piece to have it properly fitted. When resting on the diaphragm it is in focus for the upper lens, so that on looking through the microscope, the scale is clearly seen in whatever position the instrument may be as regards the object being looked at. Suppose this to be a small button of gold on a shallow, flat watch-glass, on the stage of the microscope. Bring the button under the "objective" (i.e., the nose of the microscope), which should be about a quarter of an inch above the watch-glass; then looking through the instrument, raise the tube until the button of gold, or at least some dust on the glass, comes into focus. If the button is not in the field, rest the thumbs and index fingers, using both hands, on the edge of the watch-glass, pressing lightly but steadily, and give the glass a slow, short, sweeping motion; the button will perhaps appear as an ill-defined blackness, because[Pg 134] not quite in focus. Bring this into the centre of the field. Raise or lower the microscope until the button appears with sharp outlines. If the scale does not cover the button, rotate the eye-piece; this will bring the scale into a new position. Since the divisions over the button are less distinct than the others, it is best to read the latter. Thus, in fig. 44b, there are 36 divisions on one side of the button, and 35 on the other, making altogether 71. The whole scale is 80, therefore the diameter of the button is 9 divisions. The value of each division obviously varies with the magnifying power employed. With most microscopes there is a telescopic arrangement whereby the tube may be lengthened; if this be done and the button again brought in focus, it will be seen that, as measured on the scale, the button is much larger than before. It is evident, therefore, the micrometer must always be used in the same way. The method given in the appendix (page 440), for finding the value of the scale when gold buttons are to be measured is easy and satisfactory. When the button of gold is so small that there is considerable risk of losing[Pg 135] it in transferring to a watch-glass, it may be measured on the cupel, but for this purpose it must be well illuminated; this is best done by concentrating light on it with a lens, or with what comes to the same thing, a clean flask filled with water.
Most assayers, however, using a micrometer in this way, would like to know its absolute value. To do this, a stage micrometer must be purchased. This is like an ordinary microscope slide (fig. 44a, C), and when looked at through a microscope it shows (fig. 44c) lines ruled on the glass at distances of tenths and hundredths of a millimetre, ten of each, so that the full scale is 1.1 mm. In the case illustrated, 60 divisions of the scale in the eye-piece are just equal to the 1.1 mm., therefore 1 division equals .0183 mm. A cube of this diameter would contain (.0183×.0183×.0183) .0000061285 cubic mm. The corresponding sphere is got by multiplying by .5236; this gives .000003209 cb. mm. The weight of 1 cb. mm. of water is 1 milligram; and, since gold is 19.2 times as heavy as water (sp. g. = 19.2), the contents in cb. mm. must be multiplied by 19.2. This gives .0000616 milligram as the weight of a sphere of gold measuring 1 division.
If every result had to be calculated in this way the method would be very laborious; but, having the figures for the first division, those of the others may be calculated by multiplying by the cube of[Pg 136] the corresponding number. Thus, for the third division (3×3×3 = 27), the content of the cube (.0000061285×27) is .0001655 cb. mm.; the content of the sphere (.000003209×27) is .0000866 cb. mm.; and the corresponding sphere of gold (.0000616×27) is .00166 milligram. With the help of a table of cubes the whole calculation for 25 or 30 divisions may be made in half an hour, and the results preserved in the form of a table will simplify all future work.
Assay Operations.—The actual work of the assay resolves itself into three operations:—(1) The fusion of the ore and concentration of the "fine metal" (i.e., gold and silver) in a button of lead; (2) The cupellation of the lead, whereby a button of fine metal is obtained; and (3) the "parting" of the gold which separates it from the accompanying silver. The following description takes the order as here given, but the student, in learning the method, should first practise cupellation if he has not already done so; next he should practise the separation of gold from silver, taking known weights of fine gold (p. 63), varying from .5 or .3 gram down to quite minute quantities, and not resting satisfied until a sensitive balance can barely distinguish between the weights of gold taken and found. It may be noted here that if he has not a flatting mill at his disposal, then for large buttons it is better to make an alloy with eight or nine parts of silver to one of gold, and attack it with acid without previous flattening, rather than accept the risk and labour of beating out a less easily attacked alloy to the necessary thinness with a hammer. It is only after a sense of security in gold parting has been acquired, that the attack of an ore can be profitably accomplished, and even then simple and easy ores should be first taken, passing on to others more difficult, either because of a more complex mineral composition or a difficulty in sampling.
Concentration of the fine Metal in Lead.—The best flux for quartz, which makes up the earthy matter of most gold ores, is soda, and this is best added as carbonate or bicarbonate. By theory,[20] 50 grams of quartz will require 88.5 grams of the carbonate, or 140 grams of the bicarbonate, to form sodium silicate, which is a glassy, easily-fusible substance, making a good slag. If the bicarbonate is used, and heat is applied gradually, steam and carbonic acid are given off at a comparatively low temperature, and the carbonate is left; at a higher temperature (about 800° C., or a cherry-red heat) the carbonate fuses attacking[Pg 137] the quartz, and giving off more carbonic acid; as the heat increases, and the attack on the quartz (which of itself is infusible) becomes complete, the whole mass settles down to a liquid sodium silicate, which is sufficiently fluid to allow the gold and lead to settle to the bottom. The fluid slag does to a certain extent dissolve some of the crucible, but not seriously. In a perfect working of this experiment, the first evolution of gases (steam and carbonic acid) should be gentle, so as to run no risk of its blowing the fine powder out of the crucible; and the heat at which the second evolution of carbonic acid is produced should be maintained until the reaction is completed, so that there may be little or no formation of gas in the fused mass to cause an effervescence which may force some of the charge over the edges of the crucible. Of course, in practice the ideal fusion is not attained, but there is no difficulty in approaching it closely enough to prevent the charge at any time rising above the level it reached at first in the crucible, and this should be accomplished. It is usual with quartzose ores to rely mainly on the action of carbonate of soda, but not entirely. Litharge is also used; it forms, on fusion with quartz, a silicate of lead, which is a yellow glass, easily fusible, and more fluid in the furnace than silicate of soda is. By theory, 50 grams of quartz would require 186 grams of litharge.[21] The reaction takes place without evolution of gas, and in its working the only point is to so regulate the heat that the litharge shall not fuse and drain under the unattacked quartz, leaving it as a pasty mass on the surface. Now, if in making up a charge for 50 grams of ore, we took 100 grams of bicarbonate of soda (equivalent to about 63 grams of the carbonate), this being five-sevenths of 140 grams (which by itself would be sufficient), leaves two-sevenths of the quartz to be fluxed by other reagents: two-sevenths of 186 grams (say 52 grams) of litharge would serve for this purpose. But if we used 10 grams of borax, which has a fluxing action about equal to that of the litharge, then 40 grams of the latter, or (making an allowance for the quartz being not quite pure) say 35 grams, will suffice. The fluxes, then, for the 50 grams of ore would be: bicarbonate of soda 100 grams, litharge 35 grams, and borax 10 grams; we could decrease any of these, and proportionately increase either or both of the others, and still rely on getting a fusible slag, which is the whole of the function of a flux, considered simply as a flux. It should be remembered, however, that the slag is a bi-silicate or acid slag, and that its acid character is increased by increasing the proportion of borax.
But in addition to the fluxes there is required about 30 or 40 grams of lead to collect the silver and gold. This is best added as litharge (say 40 grams) and flour (4 grams), or charcoal powder (2 grams). See pages 93 and 94. The full charge, then, would be:
| Ore | 50 | grams. |
| Bicarbonate of soda | 100 | " |
| Litharge | 75 | " |
| Borax | 10 | " |
| Flour | 4 | " |
These should be mixed, placed in a suitable crucible (a G Battersea, round, will do), and heated, at first at a red heat, but finally much hotter, so as to get a fluid and clean slag. When the charge has been in tranquil fusion for some little time, take it out and pour it into an iron mould. When cold, detach the button of lead. The slag should be glassy, all through alike, and easily separable from the metal. With ordinary ores, this slag may be considered as free from gold. In an experiment in which 90 milligrams of gold were added, the full amount was obtained from the lead produced by the first fusion. But in certain cases, more especially where large amounts of metallic oxides are present, the slag is not so clean, and with these the slag should be powdered, mixed with 40 grams of litharge and 4 of flour, and melted again; it is an advantage to add a small prill of say 2 or 3 milligrams of silver to the charge, as it insures a visible product in the cupellation. Indeed, this last precaution is a good one to be taken wherever there is reason to expect very small buttons. It has the further advantage, that, if the quantity of silver necessary for inquartation is known, the right quantity may be added here, so as to save a subsequent operation.
Ores containing Oxides of Iron.—Of the metallic oxides likely to be present in a slag, oxide of iron is the most important. Gold is occasionally found in a matrix of this substance, and in the assay of "concentrates" largely made up of pyrites, this oxide will be formed in the preliminary calcination. Now, the lower oxide of iron (ferrous oxide, FeO) is easy to deal with; fused borax will dissolve about its own weight of it, and a silicate of soda (such as makes up the bulk of a slag in a gold assay) will take up at least half as much. But the higher oxide (ferric oxide, Fe2O3) is more refractory; even 6 parts of borax yields a poor product, and slags with any considerable percentage of it are not satisfactory. A student attempting to recover gold from some hæmatite (in which there was about half an ounce of the metal), found in the slag nearly a gram of gold, although in the first[Pg 139] fusion the slag appeared perfectly fluid. There is, however, no difficulty in getting good slags, even with large quantities of iron. For example, with 50 grams of ferric oxide, 10 of quartz, 30 of borax, 30 of soda,[22] 50 of litharge, and 7 of flour, the result was quite satisfactory. So, too, was 25 of quartz, 50 of soda, 50 of litharge, and 7 of flour. It is well, however, in such cases to have an ample proportion of flux and to aim at a larger button of lead than usual by increasing the proportion of flour or charcoal (see also page 91). A charge used on the Randt for roasted "concentrates" (which we may roughly speak of as quartz and ferric oxide), is one assay ton (about 30 grams) each of ore, soda, and borax, and one and a half assay ton of litharge and 2 grams of charcoal. Whilst, for the same material, from which most of the gold has been extracted by "chloridising," 2.5 tons each of ore, borax, and soda, 4 of litharge, and 4 grams of charcoal are needed. This quantity requires a large crucible (I Battersea, round). In this the proportion of silicate of soda and borax counted together is to the oxide of iron as 4 to 1, on the supposition that the quartz and oxide of iron of the ore are in about equal quantities; but, in the larger charge especially, much oxide of lead would also remain as a flux.
Ores containing Sulphides.—In assaying ores containing a large proportion of pyrites or mispickel, or both, the best plan is to take a portion and calcine so as to convert it into a product of the kind just considered. The weighed portion of ore should be placed in a clean crucible and be heated to incipient redness: with pyrites the first effect is to drive off about half the sulphur as vapour which burns as flame over the ore. At this stage care should be taken that there is no great increase of temperature, otherwise there may be more or less fusion, which would spoil the operation. When the sulphur flame ceases the solid sulphide of iron burns with visible incandescence and the charge should now be stirred with a flattened iron rod so as to expose fresh portions to the air. The top of the furnace must be open, so that air may have free access to the crucible. When stirring is no longer followed by visible burning the heat may be raised to full redness. The crucible is then lifted out (the stirrer still resting in it) and if the charge gives off no odour of burning sulphur it is shaken out into an iron mortar and mixed with the fluxes, taking care to clean the stirrer in the mixture. The charge is then replaced in the crucible in which the roasting was done and fused in the furnace. The resulting button of lead is cupelled for fine metal. Ores rich in sulphides requiring this treatment are frequently[Pg 140] "concentrates." For their assay take 1 assay ton (30 grams), calcine and mix with an equal weight of soda and of borax (30 grams each), and half as much again of litharge (1.5 tons or 45 grams), and with 2 grams of charcoal or 5 grams of flour.
Where the sulphides are present in smaller proportion (10 per cent. or less), they may be taken as serving the purpose of flour or charcoal (see page 95); the sulphur and iron are oxidised at the expense of the litharge with a consequent separation of lead as metal. If the proportion of sulphides is not sufficient to give a large enough button of lead, some charcoal or flour should be added. On the other hand, if they are in small excess and give a button of lead somewhat sulphury, i.e., hard and brittle, it may be remedied by the judicious addition of nitre; this last reagent, however, should not be used in large quantity. A plan much used to prevent sulphury buttons is to insert an iron rod or a nail in the charge in the crucible; the iron takes the sulphur forming sulphide of iron which in moderate quantity does not form a separate layer of matte but dissolves in the slag. A slag formed of 50 grams of quartz, 100 soda, and some borax, may take up in this way some 10 or 12 grams of sulphide of iron. If, however, an ore gives a layer of matte or speise, it is best to repeat the assay by the method of calcining before fusion.
Cyanide Charges, etc.—In assaying the "tailings" which are to be treated in a cyaniding plant the following charge is used:
| Tailings | 3 | assay tons or | 100 | grams. |
| Litharge | 4.5 | " | 150 | " |
| Soda | 4.5 | " | 150 | " |
| Borax | .75 | " | 25 | " |
The sand is assayed without any further crushing and the assay is made in duplicate.
The residues after treatment with cyanide, differing from the tailings merely in being poorer in gold because of the extraction by the solution of cyanide, are run down with the same fluxes in the same relative proportions. But four charges of 2.5 assay tons (say 75 grams) are worked, and two of the resulting buttons are scorified together and then cupelled, etc., so as to give duplicate assays on charges of 5 assay tons. This is one of the cases in which it is desirable to add a small portion of silver before cupelling.
In assaying the "cyanide liquors" for gold, 2 assay tons of the liquor are measured out (58.3 c.c. for the ton of 2000 lbs., 65.3 c.c.[Pg 141] for the other) and are evaporated to dryness in a lead dish weighing about 35 grams. Such a dish is easily extemporised out of a piece of lead foil, if the ordinary vessel is not at hand; but care must be taken that the lead is free from gold. The dish with the dried residue is then scorified and the resulting button of lead is cupelled.
In some cases the fusion of the ore may be replaced by a treatment with solution of cyanide of potassium and the gold recovered from the solution in the way just described. For this purpose the ore should be in not too fine powder, otherwise there will be great difficulty in filtering; a sand which will pass a 30 sieve and having no large proportion of very fine stuff will do. Not less than 200 grams should be taken; and as an extraction apparatus a bell jar capable of holding half as much again may be used. Such a jar may be extemporised by cutting off the bottom of a bottle by leading a crack around it with a red hot poker; or a lamp chimney will serve the purpose. The smaller mouth of the jar is closed by a perforated cork provided with a clipped tube after the manner of a burette (see fig. 44d). In the jar, just over the cork, put a plug of loose asbestos or glass wool, or a piece of sponge to act as a filter; a layer of broken glass, coarse at the bottom and fine at the top, will serve the same purpose. On this, place the charge of ore to be extracted. Prepare a solution of cyanide of potassium in water, with 5 or 10 grams of the salt to the litre. It may be that the whole point of the assay depends on the solution being of a definite strength; as, for example, where the relative efficiency of solutions of different strengths is being determined, when it will be best to estimate the quantity of cyanide of potassium in the dilute solution by the method given at the end of this article (page 160). Pour the cyanide solution on to the ore, letting the first portions to come through run into the beaker, but as soon as the ore is thoroughly wetted close the clip and allow to stand for several hours. Then, opening the clip, run through more cyanide solution and then water, so as to wash the gold-carrying liquor thoroughly into the beaker. It is no matter if the liquor is a little bit turbid; transfer it to a lead dish, evaporate, scorify, and cupel in the usual fashion.[Pg 142]
The assay of gold-zinc slimes, which is the precipitate formed by zinc acting on cyanide solutions of gold, may be made by wrapping 2 or 3 grams in 40 grams of sheet lead and scorifying, cupelling, &c. The amount of impurity in the stuff varies greatly; it is usually calcined and mixed thoroughly with soda 40 per cent., borax 30 per cent., and sand 10 per cent., and melted in graphite pots. The buttons of bullion obtained are afterwards remelted with borax and run into bars, the fineness of which varies from 600 to 830 thousandths. The bars are sampled by chipping off diagonally opposite corners: or better, by drilling, the drillings being freed from pieces of steel with the help of a magnet.
Cupellation.[23]—The cupellation of lead for gold differs very little from that of lead carrying silver. When the gold is accompanied by a larger proportion of silver, and both have to be determined, the cupellation must be conducted exactly as in a silver assay, the usual precautions being taken to moderate the temperature so as to lessen the cupellation loss and to promote a slow and undisturbed solidification in order to avoid spirting. If, however, the gold predominates the finish should be effected at a higher heat, as the melting-point of gold is 100° higher than that of silver. The bad effect of a higher temperature in increasing the cupellation loss need hardly be considered in the case of such small buttons of gold as are obtained in assaying gold ores, as any loss there may be is hardly appreciable by the balance. With larger quantities of gold, however (as in assaying gold bullion), this loss becomes important; and it is therefore necessary to very carefully regulate the temperature of the muffle so as to minimise the loss.
The cupels are made of well-burnt bone-ash, of the fineness of coarse wheat flour, moistened with one-twelfth its weight of water and compressed into shape in suitable moulds. The moulds sold for this purpose are often of unsuitable shape. Since lead has a specific gravity of over 11, a cup to hold from 15 to 25 grams of molten lead need not have a capacity of more than about 2 c.c. A hollow about 1 inch across and 1/4 inch deep is sufficient; and the body of the cupel to absorb this weight of lead should itself weigh from 20 to 25 grams. The button of lead in a gold assay may be twice as heavy as this. For these larger buttons a hollow 1-1/3 inch across and 1/3 inch deep will be sufficient. If these larger cupels are not at hand the larger buttons will have to be reduced in size by a scorification before cupelling. In some cases this preliminary scorification is advantageous or even necessary: this may be because the lead is hard and impure, or it may be that a very small button of gold is expected. In the latter case it is best to[Pg 143] scorify the lead down to something less than 1 gram, and to perform the cupellation on a specially prepared small fine cupel. These small cupels are best made by grinding the unsaturated portion of a used cupel to a fine powder, and compressing the dry powder into a small Berlin crucible or scorifier; the face should be made quite smooth by pressure from a pestle. On such cupels a small speck of gold (less than .01 milligram) will be left in a good shape and easily visible; but the cupel must be withdrawn from the muffle as soon as the cupellation is finished to make sure of always getting the button in good condition. In places, such as Mints, where large numbers of bullion assays are regularly made a special form of cupel is used so that not less than six dozen assays may all be cupelled at the same time in a muffle of ordinary size. These cupels are square blocks, a little less than 2 inches across, and a little more than three quarters of an inch deep. Each block carries four hollows of about .7 inch across and .3 inch deep. A muffle, on a floor space of 6 inches by 12, would take 3 of these blocks abreast and 6 deep, and thus provide the means for 72 assays.[24]
Cupels made with wet bone-ash should be slowly dried; and if in the muffle they can be slowly brought to an orange-red heat it is all the better. Under no circumstances must the lead be placed on the cupel before the latter has been so thoroughly heated that it can no longer give off steam or gas of any kind. For this gas bubbling through the molten metal spatters it, thus spoiling one assay and throwing doubt on all the rest. Again, the risk of freezing at the start is much greater with a cupel which has not been properly heated.
The best plan is to do all the cupellations in batches. After the muffle has cooled down for the withdrawal of the last batch, and the old cupels have been taken out, the new cupels for the next batch should be put in their place. The furnace should then be stoked and made ready for the next cupellations; by the time the furnace is ready the cupels will be ready also. There should be no unnecessary handling of the cupels once they have been placed in the muffle.
The cupellation temperature for gold is an orange-red heat or perhaps a little hotter. Beginners, who are apt to overheat their furnace, should avoid a heat which can properly be called yellow. Dr. T.K. Rose[25] has determined the temperature of a muffle during the cupellation of gold-silver alloys at the Royal Mint. In one muffle the temperature ranged from 1065° to[Pg 144] 1095° C.; the lower temperature was of course in the front of the muffle. In another it ranged from 1022° to 1062°, and here the muffle appeared to the eye "decidedly cooler than usual." The alloy left after cupelling was made up of 1 part of gold to 2-1/2 parts of silver, and was fused at 952°; hence the usual temperature of cupellation was, say, 120° or 130° above the melting-point of the residual metal. To obtain some real knowledge as to the meaning of these figures, the student should prepare pointed pieces of the following metals: silver, which melts at 945°; gold, which melts at 1035°; and an alloy, half silver, half gold, which melts at 990°. These should be placed on clean cupels in a muffle almost entirely closed; the temperature should be very slowly raised, and the appearance of the muffle when each metal begins to melt should be carefully noted. The cupelling temperature in Dr. Rose's experiment was as much above the melting-point of gold as this is above that of the silver-gold alloy. The finish of the cupellation of gold or gold-silver alloys is practically the same as with pure silver; there is the same thinning out of the litharge into a luminous film which becomes iridescent before the brightening. But the danger of spirting decreases as the proportion of gold becomes greater, and disappears when the gold is much over 30 per cent. Nevertheless it is well to let such buttons become solid undisturbed and protected from draughts in the body of the muffle. This means closing the muffle and allowing the furnace to cool down somewhat before withdrawing the cupels. Buttons solidified in this way are more malleable than when they are withdrawn promptly on the finish of the cupellation. This is important with large buttons, as in a bullion assay. On the other hand, very small buttons, especially such as have to be measured rather than weighed, should be withdrawn as soon as the luminous film has disappeared. For when this is done the button can be loosened from the cupel by merely touching it with the point of a pin, and is then safely and easily transferred to a watch glass by touching it with the head of a pin which has been moistened. It adheres to this, and if the pin is not too wet comes off at once on touching the glass, or in any case will do so on gentle warming.
Molten gold, with little or no silver, has a peculiar colour which is easy to recognise; it is more globular than a button of silver of the same size would be, and it shows less adhesion to the cupel. Just after becoming solid it glows beautifully, and this is so marked that it is a valuable help in finding the position of a button when it is more than ordinarily minute.
If the button left from cupellation is yellow it is at least half gold, and a rough guess as to the proportion of gold may be made from its yellowness; the rest of the metal is generally silver. The[Pg 145] presence of platinum or one of the platinum group of metals makes the surface of the button dull and crystalline. The native alloy of osmium and iridium does not alloy with gold, however, but falls to the bottom of the molten metal. It shows itself in the subsequent parting as a black spot or streak on the under surface.
The buttons are removed from the cupel with a pair of pliers and then brushed to remove adherent litharge and bone-ash. Some assayers advise cleaning by dipping in warm dilute hydrochloric acid followed by washing in water and drying. The button is next weighed. When the quantity of silver obtained is not required to be known the weighing may sometimes be omitted. The next operation in either case is parting either with or without a previous inquartation.
The loss of gold in cupellation is by no means always inconsiderable. In three cupellations of 1 gram of gold with 20 grams of lead made purposely at a very high temperature the cupel absorbed 6.04, 6.20, and 6.45 milligrams of gold. Hence at a high temperature there may easily be a loss of more than half a per cent. of the gold. In ten cupellations with the same quantities of gold and lead, but at an ordinary temperature, the gold recovered from the cupels varied from 1.37 to 1.92 milligrams, and gave an average of 1.59 milligrams. In round numbers the cupellation loss of pure gold is .15 per cent.
But if the gold be alloyed with silver the loss is diminished, as is shown by the following experiments. Gold, .3 gram, was cupelled with 10 grams of lead and varying amounts of silver, and the cupels were assayed for gold with the following results:
| Silver in the alloy | .3 gram | .6 gram | .9 gram |
| Gold in the cupel | .47 milligram | .32 milligram | .17 milligram |
These, calculated on the .3 gram of gold, give the loss as .157, .107 and .057 per cent. respectively. The effect of copper, on the other hand, is to increase the cupellation loss, which, silver being absent, may from this cause rise to .3 per cent., even when the temperature is not excessive.
In the ordinary assay of gold-copper alloys a constant weight of the alloy is always taken; hence as the weight of copper in a cupel charge increases, the weight of gold decreases. The silver, on the other hand, is always very nearly two and a half times as much as the gold, whatever its quantity may be. But the cupellation loss is smaller with less gold and greater with more copper, and it so happens in these assays that these two opposites nearly neutralise one another. Mr. W.F. Lowe[26] found the gold recoverable[Pg 146] from the cupels on which 20 grains of gold bullion had been treated varied only between .014 and .015 grain (i.e. from .07 to .075 per cent. of the bullion treated), although the quality of the bullion varied from 9 to 22 carat.[27] But in the poorest bullion there was only 7.5 grains of pure gold, while in the richest there were 18.3 grains; yet each lost on the cupel the same weight of gold, viz., .014 grain. When reckoned in percentages of the actual gold present the losses are .187 per cent. and .076 per cent. respectively. The heavier percentage loss is mainly due to the increased quantity of copper.
As with silver so with gold the predominant cause of the cupellation loss is the solution of the metal in the molten litharge which passes into the cupel. Three lots of 1 gram of gold cupelled each with 20 grams of lead repeatedly, so as to make 13 cupellations in all, lost in actual weight 35.72 milligrams. The gold recovered from the cupels amounted altogether to 34.56 milligrams. This shows that, compared with the absorption by the cupel, the other causes of loss are inconsiderable.
The loss of gold by volatilisation is, however, a real one. The dust from the flues of assay furnaces has been tested on several occasions and found to contain gold, though in small quantity. Thus Mr. Lowe found .073 per cent. of silver and .00033 per cent. of gold in such a material. The lead volatilised from a gold bullion assay would need to be ten times as rich as this to account for a loss of gold equal to the hundredth part of a milligram. Dr. Rose, in the paper already quoted, believes that on a .5 gram charge of standard bullion the loss from volatilisation is not less than .025 nor more than .05 milligram of gold.
By way of conclusion it may be said that the cupellation loss of gold is about .07 per cent., and that it is largely met or even over corrected by a compensating error due to silver retained in the gold after parting.
Inquartation.—The method of separating the gold from the silver in gold-silver alloys by boiling with nitric acid does not act equally well in all cases. An alloy half silver half gold, rolled to thin sheet and boiled for half an hour with nitric acid, may still retain more than two-thirds of its silver. An alloy of 1 part gold and 1.7 parts of silver gives up practically the whole of its silver under similar treatment. The gold is left in a coherent, though easily broken, sheet retaining the shape of the original alloy. The gold thus left is quite spongy and porous, so that the acid can penetrate into its innermost portions. But if the silver is in[Pg 147] large excess in the alloy, the removal of the silver is less complete, and the residual gold, instead of holding together in a form easy to manipulate, falls to a powder which requires care and time in its treatment. The older assayers, therefore, added silver to their gold in such proportion that the alloy for parting should be one quarter gold to three quarters silver. This operation they called inquartation.
The modern practice is to aim at getting an alloy with 2-1/2 parts of silver and 1 part of gold. In gold bullion assays this proportion should be obtained with fair exactness. And in the parting of such gold buttons as are obtained in assaying ores it is well to aim at this proportion, though absolute precision is not a matter of importance.
If the button left on cupelling the lead from an assay of an ore appears white, it is best to assume that it already contains at least a sufficiency of silver, in the absence of any knowledge to the contrary. This will be true in almost all cases. But if, on parting, it does not lose at least two-thirds of its weight, this indicates that the assumption was not justified; and also what quantity of silver must be added to the button before again attempting to part. Generally the fault will be in the other direction; the silver will be in excess and the gold will break up and demand very careful treatment.
If, however, such a button is yellow, then, from its weight and depth of colour, a rough estimate can be made of how much gold is contained in it. Silver must be added to make the total weight 3-1/2 times as much as that of the gold supposed to be present. Thus, if the button weighs 10 milligrams and is supposed to contain 8 milligrams of gold, then 8 multiplied by 3-1/2 is 28; the button must, in such case, be made up to 28 milligrams by adding 18 milligrams of silver. In judging of the quality of the gold button, no ordinary error will very seriously affect the result. If, in the example just given, the quantity of gold present was really 7 or even 9 milligrams of gold, the resulting alloy would still have been suitable for such partings. In fact, in routine assays, where the quantity as well as the quality of the gold is known within fair limits, it is often the custom to add the silver for inquartation to the lead during the first cupellation.
But in the assay of rich gold alloys such approximate work will not do. If the composition is not already known with a fair degree of accuracy preliminary assays must be made. Weigh up two lots of 100 milligrams of the alloy and wrap each in 3 grams of lead. To one add 300 milligrams of silver. Cupel both. The button containing the added silver must be flattened and boiled with 15 c.c. of nitric acid; and the resulting gold[Pg 148] must be washed, dried, ignited and weighed. This, in milligrams, gives directly the percentage of gold. The weight of the other button gives the percentage of gold and silver; the difference between the two gives the percentage of silver. The rest will, perhaps, be copper.
The composition of the alloy being known, or having been determined as just described, the calculation of how much silver must be added is fairly simple. The following is an example. Suppose the bullion contains 92 per cent. of gold, 1 per cent. of silver and 7 per cent. of copper, and that .5 gram of it is to be taken for an assay. The .5 gram, then, will contain
| Gold | .460 | gram |
| Silver | .005 | " |
| Copper | .035 | " |
But the total silver required is .46 gram × 2.5. This equals 1.15. Allowing for the .005 gram of silver already present, 1.145 gram of silver must be added.
The silver is incorporated with the gold, and at the same time the copper is eliminated, by cupelling with sheet lead. How much sheet lead must be used will depend partly on how much bullion is taken, partly on how much copper it contains. Four grams of lead will do for a .5 gram charge; and for a .3 gram charge, 3 grams may be used. But with 20 per cent. of copper these amounts should be doubled; with 40 per cent. of copper they should be trebled; and with over 60 per cent. of copper four times as much lead should be used. For small buttons of gold as little lead as may be relied on to start cupelling may be taken; the lead may conveniently be in the form of little cups made by folding lead foil on a piece of glass rod. With a large number of bullion assays systematically worked and checked a simple plan would be to always use the quantity of lead required by the alloy containing most copper which turns up for assay. This weight, cut out of lead foil, would be kept in stock folded into little bags ready to receive the bullion and silver.
The silver used for inquartation must, of course, be free from gold and is best prepared by the assayer who is to use it (see p. 66). It should not be in long strips or angular pieces likely to perforate the lead in which it is folded. When wrapped in the lead it should be in the middle and should make as compact a parcel as possible.
Each little parcel, as completed, should be placed on a tray in its properly numbered compartment. Its position here should correspond to that it will occupy in the muffle and eventually in the cupel tray. The cupellation must be made with all the[Pg 149] requisite precautions. A good smooth malleable button is needed for the next operation, which is known as flatting.
Flatting.—Small buttons, such as are got in assaying most gold ores, are placed on a polished steel anvil and flattened by one or two blows with a hammer. The flattened discs are heated to dull redness on a clean cupel and are then ready for parting. Somewhat larger buttons may be similarly treated, but they should be annealed (i.e. heated to redness and allowed to cool) during the flattening. The silver-gold alloy left from the cupellation is soft and bends like lead; but after hammering or rolling it becomes harder, gets a spring in it like a piece of mainspring and cracks or splits somewhat easily. There should be no cracks or stripping or even roughness on the flattened metal, since such defects may cause the loss of small particles either during the flattening or in the subsequent treatment with acid. The softness of the metal is restored by heating. In bullion assays the flatting of the buttons requires care and practice for its skilful working. The strips of alloy for parting should be of uniform thickness and condition so that the action of the acid shall be equal in all cases. The button is taken from the cupel, cleaned and placed on the anvil: it is then struck a heavy blow which widens it to about 3/4 inch in diameter; this blow is followed by two others, one a little in front, the other behind, which lengthen the disc and give a very blunt roof-like slope to its upper face. It should then be annealed. This may be done by putting it in a just red-hot scorifier heated in a muffle: it very soon attains the right heat and may then be transferred to a cold scorifier; the hot scorifier should be put back into the muffle. The softened disc is then taken to the rolls (Fig. 45). The rolls are loosened until the disc can be pressed between them. Looking through the interval between them the rolls should appear exactly parallel; if they are not, one adjusting screw should be loosened and the other tightened until parallelism is obtained. The rolls are now turned and the disc should be drawn through without any great effort. Beginners are apt to err by trying to do too much with one turn of the handle. It is easy to stop whilst the rolls are only just gripping the metal and then to bring the disc back by reversing the action. If the disc was originally level[Pg 150] and the rolls are parallel, the metal will appear as a strip which has been merely lengthened. If the rolls are tighter on one side the strip will be bowed; the tighter side will correspond with the outer curve of the crescent. A mistake of this kind may be amended by passing the strip through the rolls the other way, so as to reverse the irregularity and so straighten the strip. The screw on the looser side should then be tightened until parallelism is obtained; after which more care should be taken to tighten the two screws equally. The rolling should be stopped when the strip is 3 or 4 inches long and of the thickness of an ordinary visiting card. The strip should be annealed during the rolling and again at the finish.
Parting.—The thin sheet of metal is dropped into hot dilute nitric acid and boiled for five or six minutes after the brisk action of the acid on the metal has ceased. At this stage nearly all the silver has gone into solution as nitrate of silver and the acid is charged with this salt. This acid is poured off and the residual metal is again boiled for from 20 to 30 minutes with a second lot of stronger acid. This leaves the gold almost pure, though it may still retain from .05 to .1 per cent. of silver. Treatment with the first acid only would probably leave three or four times as much.
The nitric acid used should be free from hydrochloric, sulphuric, iodic and telluric acids. In testing it for the first of these add nitrate of silver and dilute with distilled water; there should be no turbidity. In testing for the others evaporate three lots in dishes over a water-bath. Test one for sulphates by adding water and barium chloride. Test another for iodates by taking up with a little water, adding a few drops of starch paste and then dilute sulphurous acid solution a little at a time; there should be no blue colour. Test the third for tellurium by heating with 1 c.c. of strong sulphuric acid until dense fumes come off; allow to cool considerably; a piece of tin foil added to the warm acid develops a fine purple colour if only a trace of tellurium is present.
The presence of lower oxides of nitrogen, which impart a brown colour to the acid, is objectionable; they, however, are removed by boiling the diluted acid before using it for parting. It is usual to keep a stock of the acid suitably diluted to the two strengths required for the parting. These are known as the parting acids. The first parting acid is the weaker and is used in the first attack on the metal. The specific gravity generally recommended for it is about 1.2. It may be prepared either by diluting the strong acid with about its own volume of distilled water, or by suitably diluting the second parting acid which has been[Pg 151] already used in an assay; the small proportion of silver this contains is not harmful for this purpose. The second parting acid has a specific gravity of about 1.3, and may be made by diluting the strong acid with half its volume of distilled water.
Parting in Flasks.—Flasks are most convenient for the larger partings, as in bullion assays; and should always be used for this purpose unless some of the special parting apparatus, like that used in Mints, is available. Many assayers use flasks, though of a smaller size, for the ordinary partings in assaying gold ores. The flasks are either bulbs with long necks (Fig. 46) which ought to be heated on rose burners of special construction; or they are small flat-bottomed conical flasks which may be conveniently heated on a hot-plate and are, in this respect, much easier to deal with in general work. The following instructions apply to the parting of an alloy containing a few decigrams of gold together with the proper proportion of silver.
The strip from the rolls, after being softened by annealing, is folded on itself on a glass rod into a roll or cornet. It should be so plastic that it will retain the shape thus given it and not spring open on removing the pressure of the fingers. About 50 c.c. of the first parting acid are placed in a 6-ounce conical flask and heated to boiling; the flask is then withdrawn, and tilted a little to one side, whilst the cornet is cautiously dropped into it; there will be a sudden issue of hot vapours and a prompt withdrawal of the hand is advisable. The flask is replaced on the hot plate and the acid is kept boiling for 10 or 15 minutes. The flask is then withdrawn and the acid diluted with about an equal volume of distilled water. If the flask has a thick glass band around its neck, a little way down,[28] care must be taken to use hot water, for any sudden chill will certainly crack the flask where it is thus thickened. The liquor is carefully decanted into a clean beaker and is then thrown into a jar marked "waste silver." About 40 c.c. of the second parting acid, heated to boiling, is then poured into the flask, which is then replaced on the hot plate. The boiling is continued for 15 or 20 minutes or even longer. At this stage bumping has to be specially guarded against; after a little experience it is easy to see when this is imminent and the flask should be withdrawn to a cooler part of the plate; it is better to prolong the heating at a temperature below boiling than to run the risk of disaster. Some of the older writers, however, are[Pg 152] rather insistent on vigorous boiling with large bubbles. The addition of a small ball of well-burnt clay of about the size of a pea has been recommended, as it lessens the tendency to irregular and dangerous boiling. At the end of the treatment with the second acid the flask is withdrawn from the plate and the acid is diluted with an equal volume of distilled water. The liquor is carefully decanted into a beaker, and then poured into a jar or Winchester marked "acid waste"; it serves for making the first parting acid. The flask is then washed twice with hot distilled water; the washings must be carefully decanted from the gold. The flask is then filled with water. A parting cup (size B) is then placed over its mouth, like a thimble on the tip of a finger. This cup is of unglazed porous earthenware of such texture that it absorbs the last few drops of water left on drying; and with a surface to which the gold does not adhere even on ignition. The gold should fall out cleanly and completely on merely inverting the cup over the pan of the balance. The flask and cup are then inverted so that the flask stands mouth down in the cup; a little of the water from the flask flows into the cup, but only a little. The gold falls steadily through the water into the cup. When time has been allowed for even the finest of the gold to have settled into the cup, the flask is removed. This is easiest done under water. The cup, with the flask still resting in it, is dipped under water in a basin; as soon as the neck of the flask is immersed the crucible can safely be drawn away from under it and then lifted out of the water. The flask should not be taken away first, for the rush of water from it may easily sweep the gold out of the cup. The water in the cup is then drained off and the cup is dried at not too high a temperature; for if the last drop or two of water should boil there is danger of spattering the gold out of the crucible. When it is dry, the cup is heated on a pipe-clay triangle over a Bunsen burner, or on a slab of asbestos in a muffle, to a dull-red heat. This brings the gold to "colour"; that is, the loose tender dark coloured gold becomes bright yellow and coherent; and is in a state fit to be transferred to the balance and weighed. All unnecessary transferences must be avoided. As soon as the cup is cool it may be inverted over the pan of the balance, when the gold will fall out cleanly or, at the worst, a gentle tap with the finger will be sufficient to detach it.
Parting in test-tubes, or in the smaller conical flasks, is used in the assay of gold ores of ordinary richness. The work is exactly like that just described in all its main features. Generally speaking much less acid will be used; for example, in test-tubes and for small buttons, 3 or 4 c.c. of each acid is quite enough. Again,[Pg 153] the action need not be so prolonged; 10 or 15 minutes in each acid is sufficient. So, too, the heating may be less; it is very convenient to support the test-tubes in a water-bath, or merely to rest them in a beaker of boiling water; and there is no serious objection to doing this. A smaller parting cup should be used; the A size is suitable. The button, on the other hand, should be beaten thinner than is needed for the larger partings. If the silver should be in excess and the gold becomes much broken up, ample time should be given for subsidence from the test-tube or flask into the parting cup.
Parting in glazed crucibles or dishes.—This method of working has the advantage that there is no transference of the gold until it is placed on the pan of the balance. On the other hand, in the boiling more care is required in adjusting the temperature. The following instructions apply to the treatment of very small buttons, to which the method is more particularly applicable; but very little modification is needed for the treatment of larger buttons. The smallest sized Berlin crucibles answer admirably. They should be cleaned by treatment with hot and strong sulphuric acid, followed by washing in distilled water; the comfort and ease of working mainly depends on the thoroughness of this cleaning. The crucible, one-third full with the first parting acid, is heated on the hot plate until the acid is almost boiling. The flattened and annealed button is dropped into it and the heating continued with, at most, gentle boiling for a few minutes. The crucible is then filled with distilled water, which cools it enough for easy handling; and when the gold has settled the liquor is poured off along a glass rod into a clean beaker. Any greasiness of the crucible makes itself felt here and is very objectionable. The crucible is then one-third filled with the second parting acid and the heating resumed, care being taken not to raise the temperature too high; this should be continued much longer than before, say for five or ten minutes or even longer according to the size of the button. Distilled water is again added and, when it is drained off, the washing with distilled water is twice repeated. It will not be possible to drain off the last drop of water; but if the gold is coherent, the crucible can be so inclined that this drop drains away from the gold, in which case the drying can be done rapidly; the boiling of the water will do no harm. But when the gold is much broken up, it will collect in the middle of this drop and the drying must be done gently; best by putting the crucible in a warm place. When dry, the crucible is heated till the gold changes colour, but the heat must be kept well below redness. When cold, the gold is transferred directly to the pan of the balance. With minute specks of gold which will require[Pg 154] measuring, it is best to put a small piece of lead foil (say .1 gram) in the crucible over the gold, and then heat the crucible to above redness over a blowpipe. Whilst the lead is oxidising it is easily swept round in a bath of molten litharge by merely tilting the crucible. In this way any separated specks of gold can be taken up with certainty. When the worker is satisfied that the lead has had ample opportunity for taking up the gold, the lead must be kept in one place and the heat slowly lowered. By this means the button becomes supported in comparatively pure litharge and when solid can be picked out quite easily with a pair of pliers and in a very clean condition. The lead button is then cupelled on a very fine cupel, as already described. The method of working last described destroys the crucible. If the gold is not quite so small this may be avoided. A small piece of lead foil should be hammered out until it is perfectly flexible. It is then shaped into a tray and the gold is transferred to it. The lead is then folded over, with the help of two pins; and cupelled.
If the crucible shows a black stain on heating it is because some silver remains through bad washing. It shows poor work and the assay should be repeated.
The silver retained in the gold after parting is, in bullion assays, an important matter; it is roughly equal to the loss of gold due to absorption by the cupel. Mr. Lowe working on .5 oz. of gold, obtained by parting in assaying bullion, found it to contain .123 per cent. of silver. Dr. Rose in some special assay pieces found by a less direct method of assaying, from .06 to .09 per cent. of silver. The proportion of silver retained varies in a marked way with the proportion of gold to silver in the alloy before parting. It is generally stated that the retained silver is least when this proportion is 1 to 2-1/2, and more or less silver than this leads to a less pure gold after parting.
Platinum in an alloy being parted is dissolved along with the silver either altogether or in part. It imparts a straw yellow colour to the parting acid. Palladium gives an orange colour to the acid.
The loss of gold by solution in the acid during parting is small, but easily demonstrable. On a 500-milligram charge of bullion it may amount to from .05 to .15 milligram; i.e. from .01 to .03 per cent. It is due to gold actually dissolved and not merely held in suspension.
Assaying with checks. Surcharge.—It will be seen from what has been stated that the errors in gold parting are of two kinds: viz. (1) a loss of gold on the cupel and to a less extent by solution in the acid, and (2) an apparent gain of gold due to the retention of silver in the parted material. Both errors are small,[Pg 155] and as they are of an opposite character they tend to neutralise each other. Hence they are altogether without effect on the accuracy of the assays of ores when the total gold is reckoned in milligrams. And even with the larger amounts present in bullion assays their influence is so small that an uncorrected result is still fairly accurate; the resultant error would not be more than one part in two or three thousand.
It is customary to report the purity of bullion, or its fineness as it is called, in parts per thousand of bullion. The sum of the errors of an assay, which is called the surcharge, is reported in the same way. Thus a surcharge of + .3 means that the gold as weighed was .3 part per 1000 more than the gold actually present. But a surcharge - .3 means that on the whole there was a loss of .3 part per 1000 in the assay.
Speaking roughly the retained silver will vary with the weight of gold present; if one alloy contains twice as much gold as another the retained silver will be about twice as much also. On the other hand, as already explained, the cupellation loss on the poorer alloy is as much as, or even more than, with the richer one, because of the copper, &c. present. With rich gold alloys the silver more than compensates for the loss and the surcharge is positive; but with poorer alloys the loss is greater and the surcharge is negative.
In Mints and places where bullion assays must be made with the highest attainable accuracy, the surcharge is determined by experiment, and the proper correction is made in the reports on the bullion. This is done by making assays of gold of the highest degree of purity alongside of those of the bullion whose quality has to be determined. These "checks" are so made that they do not differ from the actual assays in any material point. Thus, being of the same quality and weight and undergoing exactly the same treatment, they may reasonably be expected to have the same surcharge as the assays they imitate. Suppose the bullion being assayed varies only a little, up or down, from 900 gold and 100 copper in the thousand, and that .5 gram of it is used in each assay. A quantity of gold differing only a little from .450 gram would be very exactly weighed and placed with .050 gram of copper in the same weight of lead as is being used in the other assays. It would be cupelled, parted, &c., as nearly as possible under the same conditions as the actual assays. Suppose the pure gold weighed .45016 gram and the parted gold weighed .45025 gram, the gain in weight, .00009 gram, would be deducted from the actual assays. A surcharge correction is never applied except to bullion of the same quality as that represented by the "check assay" it was calculated from.[Pg 156]
It is evident that unless the gold is of the highest degree of purity these check assays will introduce an error almost equal to that which it is designed to remedy. Moreover, to work the checks to the greatest advantage, a very systematic and uniform method of working must be adopted.
Parting in special apparatus.—One plan for obtaining greater uniformity is to stamp each cornet with a number for purposes of identification, and to treat several, including one or more check assays in the same acid contained in a beaker; all the assays under these conditions evidently receive precisely the same acid treatment. Such a plan can of course only be adopted where there is no risk of the gold breaking up during the parting. An improvement on this is to have a porcelain basin[29] about 8-1/2 inches in diameter and with a capacity of about 1-1/2 litres. It is provided with a porcelain cover with 30 numbered holes through which tubes dip into the acid. The cover is removable. The tubes are like test-tubes and are supported by the cover; their bottoms are perforated with holes or slits. The acid is placed in the basin and boiled over a flat burner; it enters the tubes through the slits. The cornets are placed each in its proper tube. When the boiling is finished, the cover with the tubes is lifted and at the same time the acid drains back into the basin. A dip into a basin of distilled water washes at one operation all 30 assays. The cover is then put on a basin containing the stronger parting acid which is already boiling. This boiling is continued for half an hour. The cover with the 30 cornets is then lifted out from the acid and dipped two or three times in distilled water to wash off the last traces of acid. To transfer the cornets from the tubes to the porous cups the whole of the tube must be dipped under the water; otherwise the operation is exactly as when working with test-tubes.
A still simpler method of working is to use small platinum cups[30] provided with fine slits which admit the acid but retain the gold. A number of these, say 60, are supported on a platinum tray. The parting acids are boiled in platinum dishes under a hood; and the 60 cornets (each in its proper cup) are placed in the acid all at once: the tray carrying the cups is provided with a handle suitable for this purpose. After a proper boiling the tray is lifted out of the weaker acid into the stronger one, where it undergoes the second boiling. It is next dipped several times in distilled water and lastly, after a gentle drying, it is raised to an annealing temperature which must not be too[Pg 157] high for fear of the gold sticking to the platinum. After cooling, the cornets are transferred from the platinum cups directly to the pan of the balance. Here all 60 cornets have exactly the same treatment and the "checks" may be compared with great exactness with the other assays accompanying them. There is, too, a great saving of labour.[31]
Silver, &c., in gold bullion.—The base metals are generally determined by cupelling .5 gram of the alloy with 5 grams of lead. The loss in cupellation having been allowed for by any of the usual methods (see p. 104) the gold and silver contents are given. By deducting the gold the proportion of silver is obtained. The silver is generally determined by difference in this way. If it is desired to dissolve out the copper, silver, &c., and to determine them in the wet way, the gold must first be alloyed with a sufficiency of some other metal to render it amenable to the attack by acid. Cadmium is the metal generally recommended, and the alloy is made by melting together a weighed portion of the gold with five or six times its weight of cadmium in a Berlin crucible and under a thin layer of potassium cyanide.
Lead with gold or silver.—Large quantities of lead carrying gold and silver are sold to refiners in bars weighing about 100 lbs. each. The assay of these alloys presents no special difficulties, but the sampling of them is a question which may be profitably discussed.[32]
A molten metal may be conceived to have all the physical states observed in ordinary liquids, although these cannot be actually seen owing to its opaqueness. There is no doubt that pure lead at a temperature only a little above its melting-point can contain a large proportion of gold in such a manner that it may in a figurative way be spoken of as a clear solution. Any small portion withdrawn from the molten metal would afford a perfect sample. The same would be true of any pure alloy of lead and silver in which the silver does not exceed the proportion of 2-1/2 per cent.[33] On the other hand, if the molten metal contains much more than .5 per cent. of zinc, more than .1 per cent. of copper, or a larger quantity of silver, it may be likened to a turbid liquor. The resemblance holds good so far that if the molten lead be further heated, whereby its solvent power on the added metal is increased, the turbidity will disappear, or at least[Pg 158] be considerably diminished. A portion taken at random from such a molten metal may, or may not, give a good sample. The suspended insoluble matter will tend to concentrate itself in the upper or lower parts of the liquid according to whether it is heavier or lighter than it; and this separation may occur with extreme slowness or with fair rapidity. However, it is generally agreed that in the case of such alloys as occur in practice, samples taken in this way are quite satisfactory and are the best obtainable. The precautions insisted on are that the lead shall be made as hot as practicable; that it shall be stirred up at the time of taking the sample; and that the portion withdrawn shall be taken out with a ladle at least as hot as the molten metal. The further precaution that if any dross be on the surface of the metal it shall be skimmed off and separately sampled and assayed is almost too obvious to require mention. An alternative and, perhaps, better way of taking the sample is to withdraw portions at equal intervals from the stream of metal whilst the pot is being emptied; equal weights taken from these portions and mixed (by melting or in some other way) give a fair sample of the whole. In addition, separate assays of each portion will show to what extent the metal lacks uniformity in composition For example, samples taken at the beginning, middle, and end of a run gave the following results in ozs. of silver per ton: 475, 472, 466, showing an average result of 471 ozs. Fifteen fractions taken at regular intervals during the same pouring ranged from 475 ozs. to 464 ozs.: the average result was 469.8 ozs. The same lead cast into bars and sampled by sawing gave an average of 470 ozs.[34] In another case[35] samples drawn at the beginning, middle, and end of a run gave 1345 ozs., 1335 ozs. and 1331 ozs. The mean result in such cases is always a reasonably safe one, but evidently where the metal varies a good deal it is safer to take more than three dips.
Imagine such lead run into moulds and allowed to become solid as bars; the difference between bar and bar would not be greater than that between corresponding dip samples. But in each bar the distribution of the silver and gold is very seriously affected during solidification. Chips taken from the same bar of auriferous lead may show in one place 23 ozs. of gold to the ton, in another 39 ozs.; similarly with silver they may vary as much as from 900 ozs. to 1500 ozs. to the ton.
This rearrangement of the constituents of a bar takes place whilst the lead is partly solid, partly liquid. The most useful conception of such half-solidified metal is that of a felted spongy[Pg 159] mass of skeleton crystals of comparatively pure lead saturated with a still fluid enriched alloy. If the solidification of an ingot of impure tin be watched it will be evident that the frosted appearance of the surface is due to the withdrawal of the fluid portion from a mat of crystals of purer tin which have been for some time solid and a contraction of the mass. The shrinking of the last part to become solid is further shown by the collapse of the surface of the ingot where weakest; that is, a furrow is formed on the flat surface. In other cases of fused metal there is expansion instead of contraction in this final stage of the solidification, and the enriched alloy then causes the upper face of the ingot to bulge outwards. There are other causes effecting the redistribution of the metals through the ingot. There can be no general rule of wide application showing which part of a bar is richest and which poorest in the precious metals. This will depend on the quantities of gold or silver, on the quantities and kinds of other metals present and on the manner of casting. The student is advised to consult Mr. Claudet's paper which has been already referred to.
The best method of sampling such bars is to melt them all down and to take a dip sample of the molten metal in one or other of the methods already described. According to Mr. Claudet this should be done in all cases where the gold exceeds one or two ounces or where the silver exceeds 200 ozs. to the ton. If during the melting down some dross has formed this must be skimmed off, weighed and separately sampled and assayed. The clean lead also must be weighed, sampled and assayed. The mean result must be calculated. Thus 14 tons 5 cwts. of clean lead assaying 32 ozs. to the ton will contain 456 ozs. of silver; 15 cwt. dross assaying 20 ozs. to the ton will contain 15 ozs. of silver. The 15 tons of lead and dross will contain 471 ozs. of silver or 31.4 ozs. per ton.
Of the methods of sampling which avoid melting the bars, that known as sawing is the only one which is thoroughly satisfactory. In it the bars are brought to a circular saw having fine teeth and are sawn across either completely or halfway through; in this way a quantity of lead sawdust is obtained (say 1 lb. or so from a bar) which represents exactly the average of the bar along the particular cross section taken and approximately that of the whole bar. A bar of lead, which by dip assay gave 334 ozs. to the ton, gave on three transverse sections 333 ozs., 335 ozs. and 331 ozs. The variation may be greater than this, but with a large number of bars, where each bar is cut across in as far as possible a different place, these variations tend to neutralise each other and a good sample is obtained. Two or three cwt. of sawdust may be obtained[Pg 160] in this way; this is thoroughly mixed and reduced by quartering in the usual way or by a mechanical sampler. A sample of 2 or 3 lbs. is sent to the assayer. This being contaminated with the oil used in lubricating the saw is freed from it by washing with carbon bisulphide, ether or benzene and dried. Then, after mixing, 100 to 200 grams of it are carefully weighed and placed in a hot crucible, the heat of which should be sufficient to melt all the lead. The molten lead should not be overheated and should show no loss due to the melting. The removal of the oil may have decreased the weight by perhaps one half per cent. If the lead gives dross on heating it may be melted under 10 or 20 grams of potassium cyanide, which prevents the formation of dross. Samples are sometimes taken with a drill, gouge or chisel, though no method of this kind is quite satisfactory. One plan adopted is to use a punch which, when driven into the bar, gives a core or rod of metal about half as long as the bar is thick and about one-eighth of an inch across. With five bars side by side it is customary to drive in the punch at one end on the first bar, and at the opposite end on the last one, and on the others in intermediate positions in such a manner that all the holes will be along a diagonal of the rectangle enclosing the bars. The bars are then turned over and similar portions punched out through the bottoms of the bars and along the other diagonal. Or one set of five may be sampled along the top and the next set along the bottom of the bars.
Silver and gold present in bars of copper are subject to the same irregularity of distribution as in lead. The sampling of such bars is guided by the same principles.[36]
The cyanides ought perhaps to be considered along with chlorides, bromides and iodides in Chapter XV. But they are treated here because they owe their importance to their use in the extraction of gold and because their determination has become a part of the ordinary work of an assayer of gold ores.
Formerly, the cyanide most easily obtained in commerce was potassium cyanide; and it was generally sold in cakes which might contain as little as 40 per cent. or as much as 95 per cent. of the pure salt. It became customary to express the quality of a sample of commercial cyanide by saying it contained so much per cent. of potassium cyanide. The commercial product now made[Pg 161] by improved methods of manufacture is actually sodium cyanide, but is called "potassium cyanide" (probably with the words "double salt" on the label); it contains cyanide equivalent to something over 100 per cent. of potassium cyanide in addition to a large proportion of sodium carbonate and other impurities. What is wanted in most cases is merely a soluble cyanide, and it is a matter of indifference whether the base be sodium or potassium. But since 49 parts of sodium cyanide (NaCN = 49) are equivalent to 65 parts of potassium cyanide (KCN = 65) it is evident that a pure sample of sodium cyanide would contain cyanide equivalent to little less than 133 per cent. of potassium cyanide. Therefore a sample of cyanide reported on in this way may be rich in cyanide, and yet have much impurity.
The commonest impurity in commercial cyanide is carbonate of sodium or potassium. This may be tested for by dissolving, say, 2 grams in a little water and adding barium chloride. There may be formed a white precipitate of barium carbonate, which if filtered off, washed and treated with acid, will dissolve with effervescence. Cyanate may be tested for in the solution from which the barium carbonate has been filtered by adding a little soda and boiling; if cyanates are present they decompose, giving off ammonia (which may be tested for in the steam) and yielding a further precipitate of barium carbonate.[37] If the soda alone gave a further precipitate of barium carbonate, this may, perhaps, be due to the presence of bicarbonates. Alkaline sulphides may be present in small quantity in commercial cyanide. Their presence is shown at once when the sample is being tested for its strength in cyanide, inasmuch as the first few drops of silver nitrate solution produce at once a darkening of the liquor. A special test for sulphide may be made by adding a drop or two of solution of acetate of lead to four or five c.c. of soda solution and adding this to a clear solution of the suspected cyanide. This will cause a black precipitate or colour, if any sulphide is present.
The cyanides of the heavier metals combine with the alkaline cyanides to form double cyanides. Some of these, ferrocyanide and ferricyanide of potassium for example, have such characteristic properties that the fact that they are cyanides may be overlooked. Others, such as potassium zinc cyanide (K2ZnCy4), have much less distinctiveness: they behave more or less as a mixture of two cyanides and are, moreover, so easily decomposed that it may be doubted if they can exist in dilute alkaline solutions. In reporting the cyanide strength of a cyanide liquor as equivalent to so much[Pg 162] per cent. of potassium cyanide, there is a question as to whether the cyanide present in the form of any of these double cyanides should be taken into account. It must be remembered that the object of the assay is not to learn how much of the cyanide exists in the solution as actual potassium cyanide; reporting the strength in terms of this salt is a mere matter of convenience; what is really desired is to know how much of the cyanide present in the liquor is "free" or "available" for the purposes of dissolving gold. Every one is agreed as to the exclusion of such cyanides as the following: potassium ferrocyanide (K4FeCy6), potassium ferricyanide (K3FeCy6), potassium silver cyanide (KAgCy2), and potassium aurocyanide (KAuCy2); and the double cyanides with copper or nickel. But with cyanide liquors containing zinc the position is less satisfactory. One method of assay gives a lower proportion of cyanide when this metal is present; and the loss of available cyanide thus reported depends, though in a fitful and uncertain way, upon the quantity of zinc present. The other method of assay reports as full a strength in cyanide as if no zinc were present. Unfortunately, using both methods and accepting the difference in the results as a measure of the quantity of zinc present, or at any rate of the zinc present as cyanide, is not satisfactory. It appears best to use the method which ignores the zinc; and to determine the amount of zinc by a special assay of the liquor for this metal.
The cyanide present as hydrogen cyanide or prussic acid (HCy) is practically useless as a gold solvent. Hence any report on the strength of a cyanide liquor which assigned to this the same value as its equivalent of alkaline cyanide would be misleading. On the other hand, it is "available cyanide" inasmuch as a proper addition of sodium hydrate[38] would restore its value. The question of the presence or absence of free prussic acid is involved in the larger one as to whether the cyanide solution has the right degree of alkalinity. The assay for "cyanide" should include the hydrogen cyanide with the rest.
A rough test of the power of a cyanide liquor for dissolving gold may be made by floating a gold leaf on its surface and noting the time required for its solution. This test might, perhaps, be improved by taking, say, 20 c.c. of the liquor and adding three or four gold leaves so that the gold shall always be in considerable excess. The liquor should not be diluted as this will affect the result. It should be allowed to stand for a definite time, say at least two or three hours, or better, that corresponding to the time the liquor is left in contact with the ore in actual practice. The[Pg 163] liquor should then be filtered off and, with the washings, be evaporated in a lead dish as in the assay of cyanide liquors for gold (p. 141). The gold obtained on cupelling, less any gold and silver originally present in the liquor, would be the measure of the gold dissolving power.
The determination of the quantity of a cyanide is made by finding how much silver nitrate is required to convert the whole of the cyanide into potassium silver cyanide[39] or one of the allied compounds. It will be seen from the equation that 170 parts by weight of silver nitrate are required for 130 parts by weight of potassium cyanide. As already explained it is customary to report the cyanide-strength in terms of potassium cyanide, even when only the sodium salt is present. One gram of potassium cyanide will require 1.3076 gram of silver nitrate. The standard solution of silver nitrate is made by dissolving 13.076 grams of silver nitrate in distilled water and diluting to 1 litre; 100 c.c. of such a solution are equivalent to 1 gram of potassium cyanide.[40]
The titration is performed in the usual way, running the standard solution of silver nitrate into a solution containing a known weight or volume of the material containing the cyanide. The finishing point is determined in one of two ways, both of which are largely used. In the first place, as long as there remains any free cyanide in the solution the silver nitrate will combine with it forming the double cyanide and yielding a clear solution; but as soon as all the free cyanide is used up the silver nitrate will react with the double cyanide[41] forming silver cyanide, which separates as a white precipitate and renders the solution turbid. But, in the second place, if potassium iodide is present in the solution the excess of silver nitrate will react with it,[42] rather than with the double cyanide; and silver iodide will separate as a yellowish turbidity which is easily recognised.
In working with pure solutions, the two finishing points give the same results; and this is true even when there is much difference in the degree of dilution. The finishing point with the iodide,[Pg 164] however, has an advantage in precision. Moreover, it is but little affected by variations in alkalinity, which render the other finishing point quite useless. The great difference between the two is shown when zinc is present in the solution. In this case, when working without the iodide, the first appearance of a turbidity is less distinct; the turbidity increases on standing and as a finishing point is unsatisfactory. It can be determined with precision only by very systematic working and after some experience. The turbidity is due to the separation of an insoluble zinc compound. A most important point (to which reference has already been made) is that less silver nitrate is required to give this turbidity and, consequently, a lower strength in cyanide is reported. On the other hand, as much silver nitrate is required to give the yellow turbidity due to silver iodide as would be required if no zinc were present.
Unfortunately the difference in the two titrations does not depend merely on the quantity of zinc present; as it is also influenced by the extent of dilution, the degree of alkalinity of the solution, and the quantity of cyanide present. In an experiment with .055 gram of zinc sulphate and .1 gram of potassium cyanide the difference in the two finishing points was only .1 c.c.; whereas with .4 gram of potassium cyanide, the other conditions being the same, the difference was 1.5 c.c. of standard silver nitrate. On the assumption that all the zinc was present as potassium zinc cyanide (K2ZnCy4) the difference should have been 5 c.c. in each case. Again, repeating the experiment with .4 gram of potassium cyanide, but with .11 gram of crystallised zinc sulphate, the difference was 6.5 c.c.: that is, merely doubling the quantity of zinc increased the difference by more than four times. Hence it would appear better to use the method with the iodide and make a separate assay for the zinc. But since the student may be called on to use the other method, he is advised to practice it also.
The assay without iodide.—The standard solution of silver nitrate is placed in a small burette divided into tenths of a c.c. Ten c.c. of the cyanide solution to be assayed is transferred to a small flask and diluted with water to about 70 c.c. The silver solution is then run in from the burette (with constant shaking of the flask), a little at a time but somewhat rapidly, until a permanent turbidity appears. Since 1 c.c. of the silver nitrate solution corresponds to .01 gram of potassium cyanide, it also corresponds to .1 per cent. of this salt counted on the 10 c.c. of cyanide solution taken. The titration should be performed in a fairly good uniform light. The learner should practice on a fairly pure solution of potassium cyanide at first, and this may[Pg 165] conveniently have a strength of about 1 per cent. For practice with solutions containing zinc make a solution containing 1.1 gram of crystallised zinc sulphate in 100 c.c. and slowly add measured quantities of from 1 to 5 c.c. of this to the 10 c.c. of cyanide liquor before diluting for the titration.
If a cyanide solution blackens on the addition of the silver nitrate it contains sulphide. In this case, shake up a considerable bulk of the liquor with a few grams of lead carbonate, allow to settle and make the assay on 10 c.c. of the clear liquor.
If the cyanide liquor be suspected to contain free prussic acid, take 10 c.c. for the assay as usual; but, before titrating, add .1 or .2 gram of sodium carbonate. On no condition must caustic soda or ammonia be added. The difference between the results, with and without the addition of carbonate of soda, is supposed to measure the quantity of free prussic acid. If this has to be reported it is best done as "prussic acid equivalent to ... per cent. of potassium cyanide." Suppose, for example, the difference in the two titrations equals 1 c.c. of standard silver nitrate; the prussic acid found would be equivalent to .1 per cent. of potassium cyanide.
The assay with iodide.—The standard solution of silver nitrate is placed in a burette divided into tenths of a c.c. Take 10 c.c. of the cyanide liquor, which should previously have been treated with white lead for the removal of sulphides if these happened to be present. Transfer to a small flask, add 3 or 4 drops of a solution of potassium iodide and 2 or 3 c.c. of a solution of sodium hydrate; dilute to 60 or 70 c.c. with water. If much zinc is present the soda may be increased to 20 or 30 c.c. with advantage. The standard solution should be run in somewhat rapidly, but a little at a time, so that the precipitate at first formed shall be small and have only a momentary existence. The titration is continued until there is a permanent yellowish turbidity. The most satisfactory and exact finish is got by ignoring any faint suspicion of a turbidity and accepting the unmistakable turbidity which the next drop of silver nitrate is sure to produce. This finishing point gives results which are exactly proportional to the quantity of cyanide present; and it can be recognised with more than ordinary precision even in solutions which are not otherwise perfectly clear.
Each c.c. of the standard silver nitrate solution corresponds to .01 gram of potassium cyanide; and if 10 c.c. of the liquor are taken for assay this corresponds to .1 per cent. or 2 lbs. to the short ton or 2.24 lbs. to the long ton. As already explained the result should be reported as "cyanide equivalent to so much per cent. of potassium cyanide."[Pg 166]
The following experimental results were obtained with a solution of potassium cyanide made up to contain about 1.2 per cent. of the salt.
Effect of varying cyanide.—The bulk before titration was in each case 60 c.c.; 2 c.c. of soda and 3 drops of potassium iodide were used in each case.
| Cyanide added | 40 c.c. | 30 c.c. | 20 c.c. | 10 c.c. | 5 c.c. | 1 c.c. |
| Silver required | 47.0 c.c. | 35.25 c.c. | 23.5 c.c. | 11.7 c.c. | 5.8 c.c. | 1.15 c.c. |
Accepting the result for 40 c.c. as correct, the others are in very satisfactory agreement.
Effect of varying dilution.—The conditions were those of the 40 c.c. experiment in the last series; but varying amounts of water were used in diluting.
| Water added | none | 100 c.c. | 200 c.c. | 400 c.c. |
| Silver required | 47.0 c.c. | 47.0 c.c. | 47.0 c.c. | 47.05 c.c. |
Very considerable dilution therefore has no effect.
Effect of varying soda.—The conditions were those of the 40 c.c. experiment in the first series, except that varying amounts of soda solution were used.
| Soda added | none | 10 c.c. | 30 c.c. |
| Silver required | 46.95 c.c. | 47.0 c.c. | 47.0 c.c. |
This alkali therefore has no prejudicial effect.
Effect of ammonia.—Soda causes turbidity in some cyanide liquors; with these it should be replaced by 2 or 3 c.c. of dilute ammonia with a gram or so of ammonium chloride. The following experiments with dilute ammonia show that larger quantities of this reagent must be avoided.
| Ammonia added | none | 10 c.c. | 30 c.c. | 60 c.c. |
| Silver required | 46.95 c.c. | 47.15 c.c. | 47.7 c.c. | 49.5 c.c. |
Effect of sodium bicarbonate.—In this experiment 1 gram of bicarbonate of soda was used instead of the soda or ammonia of the other experiments. The silver nitrate required was only 46.45 c.c. instead of the 47.0 c.c. which is the normal result. This is probably due to the liberation of prussic acid and shows the importance of having the solution alkaline.
Effect of zinc.—In each experiment 40 c.c. of the cyanide solution and .5 gram of zinc sulphate crystals were used and the bulk was made up to 100 c.c. before titrating.
| Soda added | 1 c.c. | 5 c.c. | 10 c.c. | 25 c.c. |
| Silver required | 47.1 c.c. | 47.0 c.c. | 46.9 c.c. | 46.9 c.c. |
The work was easier with the more alkaline solutions. The titration in the presence of zinc is comparatively easy, but, in[Pg 167] learning it, it is well to have a burette with cyanide so that if a titration be overdone it can be brought back by the addition of 1 or 2 c.c. more cyanide and the finish repeated; a quarter of an hour's work in this way will ensure confidence in the method.
Effect of other substances.—It was found that an alkaline cyanate, sulphocyanate, ferrocyanide, nitrite, borate, silicate or carbonate has no effect. The ferricyanide had a small influence and, as might be expected, hyposulphite is fatal to the assay. The addition of salts of lead and cadmium was without effect. On the other hand, nickel produces its full effect; and the quantity of nickel added can be calculated with accuracy from the extent of its interference with the titration.
Assay of commercial cyanide of potassium.—Break off 20 or 30 grams of the cyanide in clean fresh pieces, weigh accurately to the nearest centigram. Dissolve in water containing a little sodium hydroxide; transfer to a 2-litre flask: dilute to 2 litres; add a few grams of white lead; shake up and allow to settle. Run 50 c.c. of the clear liquor from a burette into an 8 oz. flask; add 2 or 3 c.c. of soda solution and 3 drops of potassium iodide. Titrate with the standard solution of silver nitrate. The percentage may be calculated by multiplying the number of c.c. used by 40 (50 c.c. is one fortieth of the 2 litres) and dividing by the weight of commercial cyanide originally taken.
Alkalinity of commercial potassium cyanide and of cyanide solutions.—Hydrocyanic acid like carbonic acid has no action on methyl-orange;[43] hence the alkaline cyanides may be titrated with "normal acid" as easily as the carbonates or hydrates. 100 c.c. of normal acid will neutralise 6.5 grams of pure potassium cyanide.[44] A solution of commercial cyanide prepared as for the assay last described, but best without the addition of white lead, may be used for the test. Take 50 c.c. of it; tint faintly yellow with methyl-orange and titrate with normal acid till the liquor acquires a permanent reddish tint. In the case of the purer samples of cyanide the quantity of acid used will correspond exactly with that required to neutralise the actual quantity of cyanide present as determined by the assay with nitrate of silver. The less pure samples will show an excess of alkalinity because of the presence of sodium carbonate or of potassium carbonate.
In comparing the alkalinity and cyanide strength of a solution the simplest plan is to take 65 c.c. of the solution and titrate[Pg 168] with normal acid; for in this case each c.c. of normal acid corresponds to .1 per cent. of potassium cyanide. In systematic assays of this kind, the alkalinity would no doubt be generally in excess of that required by the cyanide present: there would be no inconvenience in recording such excess in terms of potassium cyanide.
Determination of the acidity of an ore.—Most ores have the power of destroying more or less of the alkalinity of a cyanide solution and in a proportionate degree of damaging its efficiency. An assay is needed to determine how much lime or soda must be added for each ton of ore in order to counteract this. Whether this acidity should be reported in terms of the lime or of the soda required to neutralise it will depend on which of these reagents is to be used in the actual practice. Again, if the ore is washed with water before treating with cyanide on the large scale, then the assay should be made of the acidity of the ore after a similar washing.
The standard solutions of acid and alkali used for this determination may be one-fifth normal. 200 c.c. of the normal solution should be diluted to 1 litre in each case, 1 c.c. of the resulting solutions would be equivalent to 8 milligrams of soda (NaHO) or 5.6 milligrams of lime, CaO. It must be remembered this refers to the pure bases in each case. Suppose it is desired to report as so many lbs. of lime to the short ton (2000 lbs.) of ore. Since 1 c.c. of the standard solution is equivalent to 5.6 milligrams of lime, if we take 2000 times this weight of ore (i.e. 11,200 milligrams or 11.2 grams) for the assay, each c.c. of standard solution will be equivalent to 1 lb. of lime to the short ton.[45]
Total acidity.—Weigh out 11.2 grams of the ore, place them in a four-inch evaporating dish and measure on to it from a burette 10 or 20 c.c. of the standard solution of soda. Stir the soda solution into the ore and allow to stand for 15 or 20 minutes with occasional stirring. Stir up with 30 or 40 c.c. of water, float a piece of litmus paper on the liquid and titrate with the standard solution of acid. If the ore is strictly neutral the quantity of "acid" required to redden the litmus will be the same as the quantity of "soda" originally used. If the ore is acid, less acid will be used. For example, if 10 c.c. of soda were used and only 7 c.c. of acid were required, the ore will have done the work of the remaining 3 c.c. of acid. And the ton of ore will require 3 lbs. of lime to neutralise its acidity.[Pg 169]
Acidity after washing.—Take 11.2 grams of the ore; wash thoroughly with water and immediately treat the residue, without drying, exactly as just described.
Examination of cyanide solutions for metals, &c.—Take a measured quantity of the solution, say 20 c.c.[46] and evaporate in a small dish with, say, half a c.c. of strong sulphuric acid. Evaporate at first, on a water-bath in a well ventilated place, but finish off with a naked Bunsen flame, using a high temperature at the end in order to completely decompose the more refractory double cyanides. Allow to cool; moisten with strong hydrochloric acid; warm with a little water and test for the metals in the solution by the ordinary methods. Since the quantities of the metals likely to be present may be given in milligrams the work must be carefully performed. It may be worth while to determine the proportions of lime and magnesia as well as those of the metals proper.
Or the 20 c.c. of cyanide liquor may be evaporated with 5 c.c. of strong nitric acid to dryness and gently ignited and the residue taken up with 2 or 3 c.c. of strong hydrochloric acid.
Copper, iron, and zinc can be rapidly determined in such a solution, as follows. Dilute with water to 10 or 15 c.c., add an excess of ammonia, and filter. The precipitate will contain the iron as ferric hydrate; dissolve it in a little hot dilute sulphuric acid: reduce with sulphuretted hydrogen; boil off the excess of gas, cool and titrate with standard potassium permanganate (p. 236). Determine the copper in the filtrate colorimetrically (p. 203); but avoid further dilution. Then add dilute hydrochloric acid, so as to have an excess of 4 or 5 c.c. after neutralising the ammonia; add some clean strips of lead foil, and boil until the solution has for some time become colourless. Titrate with standard potassium ferrocyanide (p. 263) without further dilution, and bearing in mind that at most only one or two c.c. will be required.
Examination of an ore for "cyanicides."—Place 100 grams of the ore with 200 c.c. of a cyanide solution of known strength (say .1 or .2 per cent.) in a bottle and agitate for a definite time, such as one or two days. Filter off some of the liquor and assay for cyanide, using say 20 c.c. Calculate how much cyanide has been destroyed in the operation. Evaporate 20 c.c. with sulphuric or nitric acid and examine for metal. Test another portion for sulphides, &c.
The student who has mastered the methods of assaying can greatly improve himself by working out such problems as the above.[Pg 170]
Platinum occurs in nature in alluvial deposits associated with gold and some rare metals, generally in fine metallic grains, and, occasionally, in nuggets. It is a grey metal with a high specific gravity, 21.5 when pure and about 18.0 in native specimens. It is fusible only at the highest temperature, and is not acted on by acids.
It is dissolved by warm aqua regia, forming a solution of "platinic chloride," H2PtCl6. This substance on evaporation remains as a brownish red deliquescent mass; on drying at 300° C. it is converted into platinous chloride, PtCl2, and becomes insoluble, and at a higher temperature it is converted into platinum. All platinum compounds yield the metal in this way. Platinic chloride combines with other chlorides to form double salts, of which the ammonic and potassic platino-chlorides are the most important.
Platinum alone is not soluble in nitric acid; but when alloyed with other metals which dissolve in this acid it too is dissolved; so that in gold parting, for example, if platinum was present, some, or perhaps the whole of it would go into solution with the silver. Such alloys, however, when treated with hot sulphuric acid leave the platinum in the residue with the gold.
Platinum is detected when in the metallic state by its physical characters and insolubility in acids. In alloys it may be found by dissolving them in nitric acid or in aqua regia, evaporating with hydrochloric acid, and treating the filtrate with ammonic chloride and alcohol. A heavy yellow precipitate marks its presence.
The assay of bullion, or of an alloy containing platinum, may be made as follows: Take 0.2 gram of the alloy and an equal weight of fine silver, cupel with sheet lead, and weigh. The loss in weight, after deducting that of the silver added, gives the weight of the base metals, copper, lead, &c. Flatten the button and part by boiling with strong sulphuric acid for several minutes. When cold, wash, anneal, and weigh. The weight is that of the platinum and gold. The silver may be got by difference. Re-cupel the metal thus got with 12 or 15 times its weight of silver, flatten and part the gold with nitric acid in the usual way (see under Gold), and the platinum will dissolve. The gold may contain an alloy of osmium and iridium; if so, it should be weighed and treated with aqua regia. The osmiridium will remain as an insoluble residue, which can be separated and weighed. Its weight deducted from that previously ascertained will give the weight of the gold.[Pg 171]
When the platinum only is required, the alloy must be dissolved by prolonged treatment with aqua regia, the solution evaporated to dryness, and the residue extracted with water. The solution thus obtained is treated with ammonic chloride in large excess and with some alcohol. A sparingly soluble[47] yellow ammonic platinum chloride is thrown down, mixed, perhaps, with the corresponding salts of other metals of the platinum group. Gold will be in solution. The solution is allowed to stand for some time, and then the precipitate is filtered off, washed with alcohol, dried, and transferred (wrapped in the filter paper) to a weighed crucible. It is ignited, gently at first, as there is danger of volatilising some of the platinum chloride, and afterwards intensely. With large quantities of platinum the ignition should be performed in an atmosphere of hydrogen. Cool and weigh as metallic platinum.
Occurs in nature alloyed with osmium as osmiridium or iridosmine, which is "rather abundant in the auriferous beach sands of Northern California" (Dana). It occurs in bright metallic scales, which do not alloy with lead, and are insoluble in aqua regia. Iridium also occurs in most platinum ores, and forms as much as two per cent. of some commercial platinum. In chemical properties it resembles platinum, but the ammonic irido-chloride has a dark red colour, and on ignition leaves metallic iridium, which does not dissolve in aqua regia diluted with four or five times its volume of water and heated to a temperature of 40° or 50° C.
The other metals of the platinum group are Palladium, Rhodium, Osmium, and Ruthenium. They differ from gold, platinum, and iridium by the insolubility of their sulphides in a solution of sodium sulphide. Palladium is distinguished by the insolubility of its iodide; and Osmium by the volatility of its oxide on boiling with nitric acid.
Mercury occurs native and, occasionally, alloyed with gold or silver in natural amalgams; but its chief ore is the sulphide, cinnabar. It is comparatively rare, being mined for only in a few districts. It is chiefly used in the extraction of gold and silver from their ores (amalgamation); for silvering mirrors, &c.
Mercury forms two series of salts, mercurous and mercuric, but for the purposes of the assayer the most important property[Pg 172] is the ease with which it can be reduced to the metallic state from either of these. Mercury itself is soluble in nitric acid, forming, when the acid is hot and strong, mercuric nitrate. Cinnabar is soluble only in aqua regia. Mercurous salts are generally insoluble, and may be converted into mercuric salts by prolonged boiling with oxidising agents (nitric acid or aqua regia). The salts of mercury are volatile, and, if heated with a reducing agent or some body capable of fixing the acid, metallic mercury is given off, which may be condensed and collected.
Mercury is separated from its solutions by zinc or copper, or it may be thrown down by stannous chloride, which, when in excess, gives a grey powder of metallic mercury, or, if dilute, a white crystalline precipitate of mercurous chloride. Nitric acid solutions of mercury yield the metal on electrolysis; and, if the pole on which the metal comes down be made of gold or copper, or is coated with these, the separated mercury will adhere thereto. It may then be washed and weighed.
The best tests for mercury next to obtaining globules of the metal are: (1) a black precipitate with sulphuretted hydrogen from acid solutions, which is insoluble in nitric acid; and (2) a white precipitate with stannous chloride.
Weigh up 5 grams, if the ore is rich, or 10 grams, if a poorer mineral. Take a piece of combustion tube from 18 inches to 2 feet long, closed at one end, and place in it some powdered magnesite, so as to fill it to a depth of 2 or 3 inches, and on that a layer of an equal quantity of powdered lime (not slaked). Mix the weighed sample of ore in a mortar with 10 grams of finely powdered lime and transfer to the tube; rinse out the mortar with a little more lime, and add the rinsings. Cover with a layer of six or seven inches more lime and a loosely fitting plug of asbestos. Draw out the tube before the blowpipe to the shape shown in fig. 47, avoiding the formation of a ridge or hollow at the bend which might collect the mercury. Tap gently, holding the tube nearly horizontal, so as to allow sufficient space above the mixture for the passage of the gases and vapours which are formed. Place the tube in a "tube furnace," and, when in position, place a small beaker of water so that it shall just close the opening of the tube. The point of the tube should not more than touch[Pg 173] the surface of the water. Bring the tube gradually to a red heat, commencing by heating the lime just behind the asbestos plug, and travelling slowly backwards. When the portion of the tube containing the ore has been heated to redness for some time the heat is carried back to the end of the tube. The magnesite readily gives up carbonic acid, which fills the tube and sweeps the mercury vapour before it. Some of the mercury will have dropped into the beaker, and some will remain as drops adhering to the upper part of the neck. Whilst the tube is still hot cut off the neck of the tube just in front of the asbestos plug (a drop of water from the wash bottle will do this), and wash the mercury from the neck into the beaker. The mercury easily collects into a globule, which must be transferred, after decanting off the bulk of the water, to a weighed Berlin crucible. The water is removed from the crucible, first by the help of filter paper, and then by exposing in a desiccator over sulphuric acid, where it should be left until its weight remains constant. It should not be warmed.
Example:—5 grams of an ore treated in this way gave 4.265 grams of mercury, equivalent to 85.3 per cent. Pure cinnabar contains 86.2 per cent.
Solution.—Since solutions of chloride of mercury cannot be boiled without risk of loss,[48] nitric acid solutions should be used wherever possible. No mercury-containing minerals are insoluble in acids; but cinnabar requires aqua regia for solution. In dissolving this mineral nitric acid should be used, with just as much hydrochloric acid as will suffice to take it up.
To separate the mercury, pass sulphuretted hydrogen in considerable excess through the somewhat dilute solution. The precipitate should be black, although it comes down at first very light coloured. It is filtered, washed, and transferred back to the beaker, and then digested with warm ammonic sulphide. The residue, filtered, washed, and boiled with dilute nitric acid, will, in the absence of much lead, be pure mercuric sulphide. If much lead is present, a portion may be precipitated as sulphate, but can be removed by washing with ammonic acetate. To get the mercury into solution, cover with nitric acid and a few drops of hydrochloric, and warm till solution is effected. Dilute with water to 50 or 100 c.c.[Pg 174]
This may be made by electrolysis. The same apparatus as is used for the electrolytic copper assay may be employed, but instead of a cylinder of platinum one cut out of sheet copper should be taken, or the platinum one may be coated with an evenly deposited layer of copper. Fix the spiral and weighed copper cylinder in position, couple up the battery, and when this has been done put the nitric acid solution of the mercury in its place.[49] The student had better refer to the description of the Electrolytic Copper Assay.
The mercury comes down readily, and the precipitation is complete in a few hours: it is better to leave it overnight to make sure of complete reduction. Disconnect the apparatus, and wash the cylinder, first with cold water, then with alcohol. Dry by placing in the water oven for two or three minutes. Cool and weigh: the increase in weight gives the amount of metallic mercury.
It must be remembered that copper will precipitate mercury without the aid of the battery; but in this case copper will go into solution with a consequent loss in the weight of the cylinder: this must be avoided by connecting the battery before immersing the electrodes in the assay solution. The electrolysed solution should be treated with an excess of ammonia, when a blue coloration will indicate copper, in which case the electrolysis is unsatisfactory. With a little care this need not happen. Gold cylinders may preferably be used instead of copper; but on platinum the deposit of mercury is grey and non-adherent, so that it cannot be washed and weighed.
Several methods have been devised: for the details of these the student is referred to Sutton's "Handbook of Volumetric Analysis."
1. The specific gravity of mercury is 13.596. What volume would 8 grams occupy?
2. If 3.169 grams of cinnabar gave 2.718 grams of mercury, what would be the percentage of the metal in the ore?
3. Pour solution of mercuric chloride on mercury and explain what happens.
4. On dissolving 0.3 gram of mercury in hot nitric acid, and passing sulphuretted hydrogen in excess through the diluted solution, what weight of precipitate will be got?
[9] Lead may be granulated by heating it to a little above the melting point, pouring it into a closed wooden box, and rapidly agitating it as it solidifies.
[10] A rod of iron placed in the crucible with the assays will decompose any regulus that may be formed.
[11] With buttons poor in silver the lowering of the temperature at this stage is not a matter of importance.
[12] 100 grams of the lead, or of its oxide, will contain from 1.5 to 2.5 milligrams.
[13] Still the precautions of having cupels well made from bone ash in fine powder, and of working the cupellation at as low a temperature as possible are very proper ones, provided they are not carried to an absurd excess.
[14] Be careful to remove the crucible before taking the bottle out of the basin of water; if this is not done the chloride may be washed out of it.
[15] 1 c.c. of this dilute acid will precipitate 8 or 9 milligrams of silver.
[16] Chlorides interfere not merely by removing silver as insoluble silver chloride, but also by making it difficult to get a good finishing point, owing to the silver chloride removing the colour from the reddened solution.
[17] These results were obtained when using ammonium sulphocyanate, and cannot be explained by the presence of such impurities as chlorides, &c.
[18] Multiply the standard by 1000, and dilute 100 c.c. of the standard solution to the resulting number of c.c. Thus, with a solution of a standard .495, dilute 100 c.c. to 495 c.c., using, of course, distilled water.
[19] HNa2AsO4 + 3AgNO3 = Ag3AsO4 + HNO3 + 2NaNO3.
[20]
SiO2 + Na2CO3 = CO2 + Na2SiO3
SiO2 + 2NaHCO3 = 2CO2 + Na2SiO3 + H2O.
[21] PbO + SiO2 = PbSiO3
[22] Here and elsewhere in this article when a flux is spoken of as soda the bicarbonate is meant.
[23] See the description of the process commencing on p. 98 and the explanatory remarks on p. 110.
[24] Percy, Metallurgy of Silver and Gold, p. 258.
[25] "Limits of Accuracy attained in Gold-bullion Assay," Trans. Chem. Soc., 1893.
[26] "Assaying and Hall-marking at the Chester Assay Office." W.F. Lowe. Journ. Soc. Chem. Industry, Sept. 1889.
[27] Fine or pure gold is 24 carat. Nine carat gold therefore contains 9 parts of gold in 24 of the alloy; eighteen carat gold contains 18 parts of gold in 24; and so on.
[28] The mouth of the flask must not have a rim around it.
[29] See "Assaying and Hall-marking at the Chester Assay Office," by W.F. Lowe. Journ. Soc. Chem. Industry, Sept. 1889.
[30] Percy, Metallurgy of Silver and Gold, p. 263.
[31] See also "The Assaying of Gold Bullion," by C. Whitehead and T. Ulke. Eng. and Mining Journal, New York, Feb. 12, 1898.
[32] Consult Percy's Metallurgy of Silver and Gold, p. 172; A.C. Claudet, Trans. Inst. Mining and Metallurgy, vol. vi. p. 29; G.M. Roberts Trans. Amer. Inst. Mining Engineers, Buffalo Meeting, 1898; J. and H.S. Pattinson, Journ. Soc. Chem. Industry, vol. xi. p. 321.
[33] Heycock and Neville, Journ. Chem. Soc., 1892, p. 907.
[34] G.M. Roberts.
[35] A.C. Claudet.
[36] "The Sampling of Argentiferous and Auriferous Copper," by A.R. Ledoux. Journ. Canadian Mining Institute, 1899.
[37] NaCNO + BaCl2 + NaHO + H2O = NH3 + BaCO3 + 2 NaCl.
[38] HCy + NaHO = NaCy + H2O.
[39] 2KCN + AgNO3 = KAg(CN)2 + KNO3.
[40] If it be desired to make a solution so that 100 c.c. shall be equivalent to 1 gram of sodium cyanide, then 18.085 grams of silver nitrate should be taken for each litre.
[41] AgNO3 + KAgCy2 = 2 AgCy + KNO3.
[42] AgNO3 + KI = AgI + KNO3.
[43] See pp. 322, 323, and 324 for a description of the methods for measuring the quantity of acid or alkali.
[44] KCN + HCl = KCl + HCN
[45] Taking 16.0 grams of ore, each c.c. = 1 lb. of soda to the short ton. The corresponding figures for the long ton are 12.544 grams for lime and 17.92 grams for soda.
[46] In which case each .01 gram of metal found equals 1 lb to the short ton of solution.
[47] 100 c.c. of water dissolves 0.66 gram of the salt; it is almost insoluble in alcohol or in solutions of ammonic chloride.
[48] According to Personne mercuric chloride is not volatilised from boiling solutions when alkaline chlorides are present.
[49] The solution should contain about 0.25 gram of mercury, and a large excess of nitric acid must be avoided.
Copper occurs native in large quantities, especially in the Lake Superior district; in this state it is generally pure. More frequently it is found in combination. The ores of copper may be classed as oxides and sulphides. The most abundant oxidised ores are the carbonates, malachite and chessylite; the silicates, as also the red and black oxides, occur less abundantly. All these yield their copper in solution on boiling with hydrochloric acid.
The sulphides are more abundant. Copper pyrites (or yellow ore), erubescite (or purple ore), and chalcocite (or grey ore) are the most important. Iron pyrites generally carries copper and is frequently associated with the above-mentioned minerals. These are all attacked by nitric acid. They nearly all contain a small quantity of organic matter, and frequently considerable quantities of lead, zinc, silver, gold, arsenic, bismuth, &c.
The copper ores are often concentrated on the mine before being sent into the market, either by smelting, when the product is a regulus or matte, or by a wet method of extraction, yielding cement copper or precipitate. A regulus is a sulphide of copper and iron, carrying from 30 to 40 per cent. of copper. A precipitate, which is generally in the form of powder, consists mainly of metallic copper. Either regulus or precipitate may be readily dissolved in nitric acid.
Copper forms two classes of salts, cuprous and cupric. The former are pale coloured and of little importance to the assayer. They are easily and completely converted into cupric by oxidising agents. Cupric compounds are generally green or blue, and are soluble in ammonia, forming deep blue solutions.[Pg 176]
That, for copper, next after those for gold and silver, holds a more important position than any other dry assay. The sale of copper ores has been regulated almost solely in the past by assays made on the Cornish method. It is not pretended that this method gives the actual content of copper, but it gives the purchaser an idea of the quantity and quality of the metal that can be got by smelting. The process is itself one of smelting on a small scale. As might be expected, however, the assay produce and the smelting produce are not the same, there being a smaller loss of copper in the smelting. The method has worked very well, but when applied to the purchase of low class ores (from which the whole of the copper is extracted by wet methods) it is unsatisfactory. The following table, which embodies the results of several years' experience with copper assays, shows the loss of copper on ores of varying produce. The figures in the fourth column show how rapidly the proportion of copper lost increases as the percentage of copper in the ore falls below 30 per cent. For material with more than 30 per cent. the proportion lost is in inverse proportion to the copper present.
| Copper present. | Dry Assay. | Margin. | Loss on 100 Parts of Copper. |
| Per cent. | Per cent. | Per cent. | |
| 100 | 98 | 2.0 | 2.0 |
| 95 | 92-1/2 | 2.5 | 2.6 |
| 90 | 87-3/8 | 2.6 | 2.9 |
| 85 | 82-3/8 | 2.6 | 3.0 |
| 80 | 77-3/8 | 2.6 | 3.2 |
| 75 | 72-3/8 | 2.6 | 3.5 |
| 70 | 67-1/2 | 2.5 | 3.6 |
| 65 | 62-1/2 | 2.5 | 3.8 |
| 60 | 57-5/8 | 2.4 | 4.0 |
| 55 | 52-3/4 | 2.3 | 4.2 |
| 50 | 47-3/4 | 2.2 | 4.4 |
| 45 | 43 | 2.0 | 4.5 |
| 40 | 38-1/8 | 1.8 | 4.6 |
| 35 | 33-1/4 | 1.7 | 4.8 |
| 30 | 28-1/2 | 1.50 | 5.0 |
| 25 | 23-1/2 | 1.50 | 6.0 |
| 20 | 18-1/2 | 1.56 | 7.8 |
| 18 | 16-1/2 | 1.53 | 8.5 |
| 16 | 14-1/2 | 1.48 | 9.3 |
| 14 | 12-5/8 | 1.40 | 10.0 |
| 12 | 10-5/8 | 1.37 | 11.4 |
| 10 | 8-3/4 | 1.28 | 12.8 |
| 8 | 6-7/8 | 1.14 | 14.3 |
| 6 | 5 | 1.05 | 17.5 |
| 5 | 4 | 1.00 | 20.0 |
| 4 | 3 | 1.00 | 25.0 |
| 3.75 | 2-3/4 | 0.97 | 26.0 |
| 3.50 | 2-9/16 | 0.94 | 27.0 |
| 3.25 | 2-5/16 | 0.91 | 28.0 |
| 3.00 | 2-1/8 | 0.87 | 29.0 |
| 2.75 | 1-15/16 | 0.82 | 30.0 |
| 2.50 | 1-3/4 | 0.77 | 31.0 |
| 2.25 | 1-1/2 | 0.72 | 32.0 |
| 2.00 | 1-5/16 | 0.66 | 33.0 |
The wet assay being known, the dry assay can be calculated with the help of the above table by deducting the amount in the column headed "margin" opposite the corresponding percentage. For example, if the wet assay gives a produce of 17.12 per cent., there should be deducted 1.5; the dry assay would then be 15.62, or, since the fractions are always expressed in eighths, 15-5/8. With impure ores, containing from 25 to 50 per cent. of copper, the differences may be perhaps 1/4 greater.
Wet methods are gradually replacing the dry assay, and it is probable that in the future they will supersede it; for stock-taking, and the various determinations required in smelting works and on mines, they are generally adopted, because they give the actual copper contents, and since it is obvious that a knowledge of this is more valuable to the miner and smelter. Moreover, the working of the dry method has been monopolised by a small ring of assayers, with the double result of exciting outside jealousy and, worse still, of retarding the development and improvement of the process.
The principal stages of the dry assay are: (1) the concentration of the copper in a regulus; (2) the separation of the sulphur by calcining; (3) the reduction of the copper by fusion; and (4) the refining of the metal obtained.
The whole of these operations are not necessary with all copper material. Ores are worked through all the stages; with mattes, the preliminary fusion for regulus is omitted; precipitates are simply fused for coarse copper, and refined; and blister or bar coppers are refined, or, if very pure, subjected merely to washing.
The quantity of ore generally taken is 400 grains, and is known as "a full trial"; but for rich material, containing more than 50 per cent. of copper, "a half trial," or 200 grains, is used.[Pg 178]
Fusion for Regulus.—The ore (either with or without a previous imperfect roasting to get rid of any excess of sulphur) is mixed with borax, glass, lime, and fluor spar; and, in some cases, with nitre, or iron pyrites, according to the quality of the ore. The mixture is placed in a large Cornish crucible, and heated as uniformly as possible in the wind furnace, gradually raising the temperature so as to melt down the charge in from 15 to 20 minutes. The crucible is removed and its contents poured into an iron mould. When the slag is solid, it is taken up with tweezers and quenched in water. The regulus is easily detached from the slag. It should be convex above and easily broken, have a reddish brown colour, and contain from 40 to 60 per cent. of copper. A regulus with more than this is "too fine," and with less "too coarse." A regulus which is too fine is round, compact, hard, and of a dark bluish grey on the freshly broken surface. A coarse regulus is flat and coarse grained, and more nearly resembles sulphide of iron in fracture and colour.
If an assay yields a regulus "too coarse," a fresh determination is made with more nitre added, or the roasting is carried further. With low class ores a somewhat coarse regulus is an advantage. If, on the other hand, the regulus is too fine, less nitre or less roasting is the remedy. With grey copper ores and the oxidised ores, iron pyrites is added.
Calcining the Regulus.—It is powdered in an iron mortar and transferred to a small Cornish crucible, or (if the roasting is to be done in the muffle) to a roasting dish or scorifier. The calcining is carried out at a dull red heat, which is gradually increased. The charge requires constant stirring at first to prevent clotting, but towards the end it becomes sandy and requires less attention. If the temperature during calcination has been too low sulphates are formed, which are again reduced to sulphides in the subsequent fusion. To prevent this the roasted regulus is recalcined at a higher temperature, after being rubbed up with a little anthracite. The roasted substance must not smell of burning sulphur when hot. It is practically a mixture of the oxides of copper and iron.
Fusion for Coarse Copper.—The calcined regulus is mixed with a flux consisting of borax and carbonate of soda, with more or less tartar according to its weight. Some "assayers" use both tartar and nitre, the former of course being in excess. The charge is returned to the crucible in which it was calcined, and is melted down at a high temperature, and, as soon as tranquil, poured. When solid it is quenched and the button of metal separated.[Pg 179]
The slag is black and glassy. The small quantity of copper which it retains is recovered by a subsequent "cleaning," together with the slags from the next operation.
The button of "coarse copper" obtained must be free from a coating of regulus. It will vary somewhat in appearance according to the nature and quantity of the impurities.
Refining the Coarse Copper.—The same crucible is put back in the furnace, deep down and under the crevice between the two bricks. When it has attained the temperature of the furnace the coarse copper is dropped into it and the furnace closed. The copper will melt almost at once with a dull surface, which after a time clears, showing an "eye." Some refining flux is then shot in from the scoop (fig. 48), and, when the assay is again fluid, it is poured. When cold the button of metal is separated.
The button of "fine" copper is flat or pitted on its upper surface, and is coated with a thin orange film; it must have the appearance of good copper. If it is covered with a red or purple film, it is overdone or "burnt." If, on the other hand, it has a rough, dull appearance, it is not sufficiently refined. Assays that have been "burnt" are rejected. Those not sufficiently fine are treated as "coarse copper," and again put through the refining operation.
Cleaning the Slags.—These are roughly powdered and re-fused with tartar, etc., as in the fusion for coarse copper. The button of metal got is separated (if big enough refined) and weighed.
The details of the process are slightly varied by different assayers: the following will be good practice for the student.
Determination of Copper in Copper Pyrites.—Powder, dry, and weigh up 20 grams of the ore. Mix with 20 grams each of powdered lime and fluor, 15 grams each of powdered glass and borax, and 5 or 10 grams of nitre. Transfer to a large Cornish crucible and fuse under a loose cover at a high temperature for from 15 to 20 minutes. When fluid and tranquil pour into a mould. When the slag has solidified, but whilst still hot, quench by dipping two or three times in cold water. Avoid leaving it in the water so long that it does not dry after removal. When cold separate the button, or perhaps buttons, of regulus by crumbling the slag between the fingers. See that the slag is[Pg 180] free from regulus. It should be light coloured when cold and very fluid when hot. Reject the slag.
Powder the regulus in a mortar and transfer to a small crucible. Calcine, with occasional stirring, until no odour of sulphurous oxide can be detected. Shake back into the mortar, rub up with about 1 gram of powdered anthracite, and re-calcine for 10 minutes longer.
Mix the calcined regulus with 10 grams of tartar, 20 grams of soda, and 3 grams of borax; and replace in the crucible used for calcining. Fuse at a bright red heat for 10 or 15 minutes. Pour, when tranquil.
As soon as solid, quench in water, separate the button of copper, and save the slag.
To refine the copper a very hot fire is wanted, and the fuel should not be too low down in the furnace. Place the crucible well down in the fire and in the middle of the furnace. The same crucible is used, or, if a new one is taken, it must be glazed with a little borax. When the crucible is at a good red heat, above the fusing point of copper, drop the button of copper into it, and close the furnace. Watch through the crevice, and, as soon as the button has melted and appears clear showing an eye, shoot in 10 grams of refining flux, close the furnace, and, in a few minutes, pour; then separate the button of copper. Add the slag to that from the coarse copper fusion, and powder. Mix with 5 grams of tartar, 0.5 gram of powdered charcoal, and 2 grams of soda. Fuse in the same crucible, and, when tranquil, pour; quench, and pick out the prills of metal.
If the copper thus got from the slags is coarse looking and large in amount, it must be refined; but, if small in quantity, it may be taken as four-fifths copper. The combined results multiplied by five give the percentage of copper.
The refining flux is made by mixing 3 parts (by measure) of powdered nitre, 2-1\2 of tartar, and 1 of salt. Put in a large crucible, and stir with a red-hot iron until action has ceased. This operation should be carried out in a well-ventilated spot.
For pure ores in which the copper is present, either as metal or oxide, and free from sulphur, arsenic, &c., the concentration of the copper in a regulus may be omitted, and the metal obtained in a pure state by a single fusion.[50] It is necessary to get a fluid neutral slag with the addition of as small an amount of flux as possible. The fusion should be made at a high temperature, so as not to occupy more than from 20 to 25 minutes. Thirty grams of ore is taken for a charge, mixed with 20 grams of cream of[Pg 181] tartar, and 10 grams each of dried borax and soda. If the gangue of the ore is basic, carrying much oxide of iron or lime, silica is added, in quantity not exceeding 10 grams. If, on the other hand, the gangue is mainly quartz, oxide of iron up to 7 grams must be added.
Example.—Twenty grams of copper pyrites, known to contain 27.6 per cent. of copper, gave by the method first described 5.22 grams of copper, equalling 26-1/8 per cent. Another sample of 20 grams of the same ore, calcined, fused with 40 grams of nitre, and washed to ensure the removal of arsenic and sulphur, and treated according to the second method, gave a button weighing 5.27 grams, equalling 26-3/8 per cent. The ore contained a considerable quantity of lead. Lead renders the assay more difficult, since after calcination it remains as lead sulphate, and in the fusion for coarse copper reappears as a regulus on the button.
The Estimation of Moisture.—The Cornish dry assayer very seldom makes a moisture determination. He dries the samples by placing the papers containing them on the iron plate of the furnace.
It is well known that by buying the copper contents of pyrites by Cornish assay, burning off the sulphur, and converting the copper into precipitate, a large excess is obtained.
Closely bound up with the practice of dry copper assaying is that of valuing a parcel of copper ore. The methods by which the valuation is made have been described by Mr. Westmoreland,[51] and are briefly as follows:—The produce of the parcel is settled by two assayers, one acting for the buyer, the other for the seller; with the help, in case of non-agreement, of a third, or referee, whose decision is final. The dry assayers who do this are in most cases helped, and sometimes, perhaps, controlled, by wet assays made for one or both of the parties in the transaction.
In the case of "ticketing," the parcels are purchased by the smelters by tender, and the value of any particular parcel is calculated from the average price paid, as follows:—The "standard," or absolute value of each ton of fine copper in the ore, is the price the smelters have paid for it, plus the returning charges or cost of smelting the quantity of ore in which it is contained. The value of any particular parcel of ore is that of the quantity of fine copper it contains, calculated on this standard, minus the returning charges. The ton consists of 21 cwts., and[Pg 182] it is assumed that the "settled" produce is the actual yield of the ore.
If at a ticketing in Cornwall 985 tons of ore containing 63.3 tons of fine copper (by dry assay) brought £2591 12s., the standard would be £83 15s. This is calculated as follows:—The returning charge is fixed at 55s. per ton of ore. This on 985 tons will amount to £2708 15s. Add this to the actual price paid, and there is got £5300 as the value of the fine copper present. The weight of copper in these 985 tons being 63.3 tons, the standard is £5300/63.3, or £83 15s. (nearly).
The value of a parcel of 150 tons of a 6 per cent. ore on the same standard would be arrived at as follows:—The 150 tons at 6 per cent. would contain 9 tons (150×6/100) of fine copper. This, at £83 15s. per ton, would give £753 15s. From this must be deducted the returning charges on 150 tons of ore at 55s. per ton, or £412 10s. This leaves £341 5s. as the value of the parcel.
At Swansea the returning charge is less than in Cornwall, and varies with the quality of the ore. This appears equitable, since in smelting there are some costs which are dependent simply on the number of tons treated, and others which increase with the richness. The returning charge then is made up of two parts, one fixed at so much (12s. 2d.) per ton of ore treated, and the other so much (3s. 9d.) per unit of metal in the ore. In this way the returning charge on a ton of ore of 8-3/4 produce would be 12s. 2d.+(8-3/4×3s. 9d.), or £2 5s.
If, for example, Chili bars, containing 96 per cent. of copper, bring £50 per ton, the standard is £71 9s. 4d. It is got at in this way. The returning charge on a 96 per cent. ore is 12s. 2d.+(96×3s. 9d.), or £18 12s. 2d. This added to £50 gives £68 12s. 2d., and this multiplied by 100 and divided by 96 (100 tons of the bars will contain 96 tons of fine copper) will give £71 9s. 4d.
The price of 100 tons of pyrites, containing 2-1/4 per cent. of copper by dry assay, would be got on this standard as follows:—The parcel of ore would contain 2-1/4 tons of copper. This multiplied by the standard gives £160 16s. 0d. From this must be deducted the returning charge, which for 1 ton of ore of this produce would be 12s. 2d. + (2-1/4 × 3s. 9d.) or £1 0s. 7d., and on the 100 tons is £102 18s. 4d. This would leave £57 17s. 10d. as the price of the parcel, or 11s. 7d. per ton. This would be on the standard returning charge of 45s. (for 8-3/4 per cent. ore); if a smaller returning charge was agreed on, say 38s., the difference in this case, 7s., would be added to the price per ton.[Pg 183]
The solubility of the ores of copper in acid has already been described, but certain furnace products, such as slags, are best opened up by fusion with fusion mixture and a little nitre.
The method of dissolving varies with the nature of the ore. With 5 grams of pyrites, a single evaporation with 20 c.c. of nitric acid will give a residue completely soluble in 30 c.c. of hydrochloric acid. If the ore carries oxide of iron or similar bodies, these are first dissolved up by boiling with 20 c.c. of hydrochloric acid, and the residue attacked by an addition of 5 c.c. of nitric. When silicates decomposable by acid are present, the solution is evaporated to dryness to render the silica insoluble; the residue extracted with 30 c.c. of hydrochloric acid, and diluted with water to 150 c.c. It is advisable to have the copper in solution as chloride. To separate the copper, heat the solution nearly to boiling (best in a pint flask), and pass a rapid current of sulphuretted hydrogen for four or five minutes until the precipitate settles readily and the liquid smells of the gas. When iron is present it will be reduced to the ferrous state before the copper sulphide begins to separate. The copper appears as a brown coloration or black precipitate according to the quantity present. Filter through a coarse filter, wash with hot water containing sulphuretted hydrogen, if necessary. Wash the precipitate back into the flask, boil with 10 c.c. of nitric acid, add soda till alkaline, and pass sulphuretted hydrogen again. Warm and filter, wash and redissolve in nitric acid, neutralise with ammonia, add ammonic carbonate, boil and filter. The copper freed from impurities will be in the solution. Acidulate and reprecipitate with sulphuretted hydrogen. When the nature of the impurities will allow it, this process may be shortened to first filtering off the gangue, then precipitating with sulphuretted hydrogen and washing the precipitate on the filter first with water and then with ammonium sulphide.
Having separated the copper as sulphide, its weight is determined as follows. Dry and transfer to a weighed porcelain crucible, mix with a little pure sulphur, and ignite at a red heat for 5 or 10 minutes in a current of hydrogen. Allow to cool while the hydrogen is still passing. Weigh. The subsulphide of copper thus obtained contains 79.85 per cent. of copper; it is a greyish-black crystalline mass, which loses no weight on ignition if air is excluded.
Copper may be separated from its solutions by means of sodium hyposulphite. The solution is freed from hydrochloric and nitric acids by evaporation with sulphuric acid; diluted to about[Pg 184] a quarter of a litre; heated nearly to boiling; and treated with a hot solution of sodium hyposulphite (added a little at a time) until the precipitate settles and leaves the solution free from colour. The solution contains suspended sulphur. The precipitate is easily washed, and under the proper conditions the separation is complete, but the separation with sulphuretted hydrogen is more satisfactory, since the conditions as to acidity, &c., need not be so exact.
Zinc or iron is sometimes used for separating copper from its solutions, but they are not to be recommended.
The separation of copper by means of a current of electricity is largely made use of, and forms the basis of the most satisfactory method for the determination of this metal. If the wire closing an electric circuit be broken, and the two ends immersed in a beaker of acidulated water or solution of any salt, the electricity will pass through the liquid, bringing about some remarkable changes. Hydrogen and the metals will be liberated around that part of the wire connected with the zinc end of the battery, and oxygen, chlorine, and the acid radicals will be set free around the other. Different metals are deposited in this way with varying degrees of ease, and whether or not any particular metal will be deposited depends—(1) on the conditions of the solution as regards acid and other substances present, and (2) on the intensity of the current of electricity used. For analytical purposes the metal should be deposited not only free from the other metals present, but also as a firm coherent film, which may afterwards be manipulated without fear of loss. This is, in the case of copper and many other metals, effected by a simple control of the conditions. It is necessary that the electrodes, or wires which bring the electricity into the solution, should be made of a material to which the deposited metal will adhere, and which will not be attacked by substances originally present or set free in the solution. They are generally made of platinum. There are various arrangements of apparatus used for this purpose, but the following plan and method of working is simple and effective, and has been in daily use with very satisfactory results for the last five or six years.
The battery used is made up of two Daniell cells, coupled up for intensity as shown in fig. 49—that is, with the copper of one connected with the zinc of the other. For eight or ten assays daily the quart size should be used, but for four or five two pint cells will be sufficient.[Pg 185]
The outer pot of each cell is made of sheet copper, and must be clean and free from solder on the inside. It is provided near the top with a perforated copper shelf in the shape of a ring, into which the inner or porous cell loosely fits. It is charged with a saturated solution of copper sulphate, and crystals of this salt must be added, and always kept in excess. When the battery is at work copper is being deposited on the inner surface of this pot.
The inner or porous pot contains the zinc rod, and is charged with a dilute acid, made by diluting one volume of sulphuric acid up to ten with water. The object of the porous pot is to prevent the mixing of the acid and copper sulphate solutions, without interrupting the flow of electricity. The copper sulphate solution will last for months, but the acid must be emptied out and recharged daily.
The zinc rods must be well amalgamated by rubbing with mercury under dilute acid until they show a uniformly bright surface. They should not produce a brisk effervescence when placed in the acid in the porous pot before coupling up.
The battery when working is apt to become dirty from the "creeping" of the copper and zinc sulphate solution. It must be kept away from the working bench, and is best kept in a box on the floor.
The connection of the battery with, and the fixing of, the electrodes may be made by any suitable arrangement, but the following is a very convenient plan. The wire from the zinc is connected by means of a binding screw with a piece of stout copper wire, which, at a distance sufficiently great to allow of easy coupling with the battery, is led along the back of a piece[Pg 186] of hard wood. This is fixed horizontally about one foot above the working bench. The general arrangement is shown in fig. 50, in which, however, for the sake of economy of space, the battery is placed on the working bench instead of on the floor. The piece of wood is one inch square and three or four feet long. It is perforated from front to back at distances of six inches by a[Pg 187] number of small holes, in which are inserted screws like that shown in fig. 51. These are known as "terminals," and may be obtained of any electrician. The head of each screw is soldered to the wire mentioned above as running along the back and as being connected with the zinc end of the battery. These terminals serve to fix the electrodes on which the copper is to be deposited. The wire from the copper end of the battery is similarly connected by a connecting screw (fig. 52) with another wire (H in fig. 53), which runs along the top of the rod and has soldered to it, at distances of six inches, cylindrical spirals of copper wire. These should project from the rod at points about half-way between the terminals already described. They may be made by wrapping copper wire around a black-lead pencil for a length of about three inches.
The rod is perforated from top to bottom with a series of small holes, one in advance of each terminal but as near it as possible. Into these short pieces of glass tube are inserted to ensure insulation. These receive the other electrodes, which are connected with the wire leading to the copper end of the battery, through the spirals, with the help of a binding screw. The figure will make this clear. (Fig. 53.)
The electrodes consist of a platinum spiral and cylinder. The spiral should have the shape shown in A, fig. 54. When in work it is passed through one of the holes fitted with glass tubes and connected with the copper end of the battery. The thickness of the wire of which it is made is unimportant, provided it is stout enough to keep its form and does not easily bend. The spiral will weigh about 8 grams. The cylinder (C, fig. 54) will weigh about 12 grams. It should have the shape shown in the figure. In working it is clamped to one of the terminals, and on it the copper is deposited. A cylinder will serve for the deposition of from 1 to 1.5 gram of copper. It is made by rivetting a square piece of foil on to a stiff piece of wire, and then bending into shape over a glass tube or piece of rounded wood. Each cylinder carries a distinctive number, and is marked by impressing[Pg 188] Roman numerals on the foil with the blade of a knife. The weight of each is carefully taken and recorded. They lose slightly in weight when in use, but the loss is uniform, and averages half a milligram per month when in daily use. The cylinders are cleaned from deposited copper by dissolving off with nitric acid and washing with water; and from grease by igniting.
The beakers, to contain the solution of copper to be electrolysed, are ordinary tall beakers of about 200 c.c. capacity, and are marked off at 100 c.c. and 150 c.c. They are supported on movable stands, consisting of wooden blocks about six inches high and three inches across. The bar of wood which carries the connecting wires and electrodes is permanently fixed over the working bench, at such a height that, with the beakers resting on these blocks, the electrodes shall be in position for working.
To fix the electrodes to the rod, remove the stand and beaker and pass the long limb of the spiral up through one of the glass tubes. Connect it with the free end of the copper spiral by means of a connecting screw (fig. 52), and then draw out and bend the copper spiral so that the platinum one may hang freely. Screw the wire of the cylinder to the terminal, and, if necessary, bend it so that the cylinder itself may be brought to encircle the rod of the spiral in the manner shown in fig. 53.
The general method of working is as follows:—The quantity of ore to be taken for an assay varies with the richness of the ore, as is shown in the following table:—
| Percentage of Copper in the Ore. | Quantity of Ore to be taken. |
| 1 to 5 | 5 grams |
| 5 to 10 | 3 " |
| 10 to 30 | 2 " |
| 30 to 50 | 1.5 " |
| 50 to 100 | 1 " |
The weighed quantity of ore is dissolved by evaporating with nitric acid and taking up with hydrochloric, as already described. Any coloured residue which may be left is generally organic matter: it is filtered off, calcined, and any copper it contains is estimated colorimetrically. Nearly always, however, the residue is white and sandy. The copper is separated from the solution as sulphide by means of a rapid current of sulphuretted hydrogen. The liquid is decanted off through a filter, the precipitate washed once with hot water and then rinsed back into the flask (the filter paper being opened out) with a jet of water from a wash bottle. Fifteen c.c. of nitric acid are added to the contents of the flask, which are then briskly boiled until the bulk is reduced to[Pg 189] less than 10 c.c. The boiling down is carried out in a cupboard free from cold draughts, so as to prevent the condensation of acid and steam in the neck of the flask. Twenty c.c. of water are next added, and the solution is warmed, and filtered into one of the beakers for electrolysis. The filtrate and washings are diluted with water to the 100 c.c. mark, and the solution is then ready for the battery. It must not contain more than 10 per cent. by volume of nitric acid.
The number and weight of the platinum cylinder having been recorded, both electrodes are fixed in position and the wooden block removed from under them. The beaker containing the copper solution is then brought up into its place with one hand, and the block replaced with the other so as to support it. All the assays having been got into position, the connecting wires are joined to the battery. If everything is right bubbles of oxygen at once stream off from the spiral, and the cylinder becomes tarnished by a deposit of copper. If the oxygen comes off but no copper is deposited, it is because the assay solution contains too much nitric acid. If no action whatever takes place, it is because the current is not passing. In this case examine the connections to see that they are clean and secure, and the connecting wires to see that they are not touching each other.
The action is allowed to go on for sixteen or seventeen hours, so that it is best to let the current act overnight. In the morning the solutions will appear colourless, and a slow stream of oxygen will still be coming off from the spiral.
A wash-bottle with cold distilled water and two beakers, one with distilled water and the other with alcohol, are got ready. The block is then removed, the spiral loosened and lowered with the beaker. The cylinder is next detached and washed with a stream of water from the wash-bottle, the washings being added to the original solution. The current from the battery is not stopped until all the cylinders are washed. After being dipped in the beaker of water and once or twice in that with the alcohol, it is dried in the water-oven for about three minutes, and then weighed. The increase in weight is due to deposited copper. This should be salmon-red in colour, satin-like or crystalline in appearance, and in an even coherent deposit, not removed by rubbing. It is permanent in air when dry, but sulphuretted hydrogen quickly tarnishes it, producing coloured films. With ores containing even very small proportions of bismuth, the deposited copper has a dark grey colour, and when much of this metal is present the copper is coated with a grey shaggy deposit.
It still remains to determine any copper left undeposited in the solution. This does not generally exceed four or five milligrams,[Pg 190] and is estimated colorimetrically. Thirty c.c. of dilute ammonia (one of strong ammonia mixed with one of water) are added to the electrolysed solution, which is then diluted up to the 150 c.c. mark with water. It is mixed, using the spiral as stirrer, and, after standing a few minutes to allow the precipitate to settle, 100 c.c. of it are filtered off through a dry filter for the colorimetric determination. Since only two-thirds of the solution are taken for this, the quantity of copper found must be increased by one-half to get the quantity actually present.
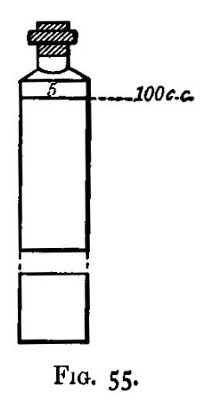
The colorimetric determination may be made in the manner described under that head, but where a number of assays are being carried out it is more convenient to have a series of standard phials containing known amounts of copper in ammoniacal solution. By comparing the measured volume of the assay solution with these, the amount of copper present is determined at a glance. These standard bottles, however, can only be economically used where a large number of assays are being made daily.
A convenient plan is to get a quantity of white glass four-ounce phials, like that in fig. 55, and to label them so that they shall contain 100 c.c. when filled up to the bottom of the labels. The labels should be rendered permanent by coating with wax, and be marked with numbers indicating the milligrams of copper present. The bottles are stopped with new clean corks, and contain, in addition to the specified quantity of copper, 6 c.c. of nitric acid and 10 c.c. of strong ammonia, with sufficient water to make up the bulk to 100 c.c. The copper is best added by running in the requisite amount of a standard solution of copper, each c.c. of which contains 0.001 gram of the metal.
The standard bottles should be refilled once every three or four months, since their colorimetric value becomes slowly less on keeping. The following determinations of a set which had been in use for three months will illustrate this. The figures indicate milligrams of copper in 100 c.c.: the first row gives the nominal and the second row the actual colorimetric value of the standards. The difference between the two shows the deterioration.
| 1 | 2 | 3 | 4 | 6 | 8 | 10 | 12 | 14 |
| 1 | 2 | 3 | 3.7 | 5.5 | 7.5 | 9 | 11 | 13 |
The amount of copper in the assay is got by increasing that found colorimetrically by one-half and adding to that found on the platinum cylinder. The percentage is calculated in the usual[Pg 191] way. The following examples will illustrate this, as well as the method of recording the work in the laboratory book:—
_____________________________________________
Cylinder I. + Cu 9.5410
Cylinder I. 9.5170
——————
0.0240
By colour 100 c.c. = 0.0015}
0.0007} 0.0022
—————— ——————
0.0022 0.0262
IX. Sample. Took 5 grams.
Copper = 0.52%
_____________________________________________
Cylinder VI. + Cu 10.5705
Cylinder VI. 10.0437
——————
0.5268
By colour, 100 c.c. = 0.0070}
0.0035} 0.0105
—————— ——————
0.0105 0.5373
Matte, No. 1070. Took 1.5 gram.
Copper = 35.82%
_____________________________________________
Cylinder XIII. + Cu 12.0352
Cylinder XIII. 11.0405
——————
0.9947
By colour 100 c.c. = 0.0005}
0.0002} 0.0007
—————— ——————
0.0007 0.9954
X. Sample, Cake copper. Took 1.0053 gram.
Copper = 99.00%
____________________________________________
In the electrolytic assay of metals, alloys, precipitates, and other bodies rich in copper, the preliminary separation of the copper by sulphuretted hydrogen is unnecessary. It is sufficient to dissolve the weighed sample in 10 c.c. of nitric acid, boil off nitrous fumes, dilute to 100 c.c. with water, and then electrolyse.
General Considerations.—In the preliminary work with the copper sulphide there is a small loss owing to its imperfect removal in washing the filter paper, and another small loss in dissolving in nitric acid owing to the retention of particles in the fused globules of sulphur. To determine its amount the filter-papers and sulphur were collected from forty assays, and the copper in them determined. The average amount of copper in each assay was 0.175 gram; that left on the filter paper was 0.00067 gram; and that retained by the sulphur 0.00003 gram; thus showing an[Pg 192] average loss from both sources of 0.00070 gram. The determinations from another lot of forty-two similar assays gave on an average
| Copper left on filter paper | 0.00070 | gram |
| Copper retained by sulphur. | 0.00004 | " |
The loss from these sources is trifling, and need only be considered when great accuracy is required.
The deposition of the copper under the conditions given is satisfactory, but, as already stated, if the solution contain more than 10 per cent. of nitric acid it is not thrown down at all; or if a stronger current is used, say that from three Bunsen cells, it will be precipitated in an arborescent brittle form, ill adapted for weighing. It may be noted here that increasing the size of the cells does not necessarily increase the intensity of the current.
In two determinations on pure electrotype copper the following results were obtained:—
| Copper Taken. | Copper Found. |
| 0.8988 gram | 0.8985 gram |
| 0.8305 " | 0.8303 " |
The presence of salts of ammonia, &c., somewhat retards the deposition, but has no other ill effect.
The organic matter generally present in copper ores interferes more especially in the colorimetric determination of the residual copper. It can be detected on dissolving the ore as a light black residue insoluble in nitric acid. It is filtered off at once, or, if only present in small amount, it is carried on in the ordinary process of the assay and separated in the last filtration before electrolysis.
The following experiments were made to test the effect of the presence of salts of foreign metals in the solution during the precipitation of copper by electrolysis:—
| Copper Taken. | Other Metal Added. | Copper Found. |
| 0.1000 gram | 0.1000 gram of silver | 0.1800 |
| 0.1050 " | 0.1000 " " | 0.2000 |
| 0.1030 " | 0.1000 " mercury | 0.2010 |
| 0.1037 " | 0.1000 " " | 0.2015 |
| 0.1020 " | 0.1000 " lead | 0.1020 |
| 0.1030 " | 0.1000 " " | 0.1028 |
| 0.1010 " | 0.1000 " arsenic | 0.1010 |
| 0.1007 " | 0.1000 " " | 0.1022 |
| 0.1030 " | 0.1000 " antimony | 0.1050 |
| 0.1034 " | 0.1000 " " | 0.1057 |
| 0.0990 " | 0.1200 " tin | 0.0990 |
| 0.1014 " | 0.1000 " " | 0.1015 |
| 0.1000 " | 0.1000 " bismuth | 0.1662 |
| 0.1040 " | 0.1000 " of cadmium | 0.1052 |
| 0.1009 " | 0.1300 " zinc | 0.1017 |
| 0.1014 " | 0.1000 " nickel | 0.1007 |
| 0.1079 " | 0.1200 " iron | 0.1089 |
| 0.1054 " | 0.1000 " chromium (Cr2O3) | 0.1035 |
| 0.1034 " | 0.1000 " " (K2CrO4) | 0.1010 |
| 0.1075 " | 0.1000 " aluminium | 0.1078 |
| 0.1010 " | 0.1000 " manganese | 0.0980 |
It will be seen from these that mercury, silver, and bismuth are the only metals which are precipitable[52] along with the copper under the conditions of the assay. Mercury, which if present would interfere, is separated because of the insolubility of its sulphide in nitric acid.
Bismuth is precipitated only after the main portion of the copper is thrown down. It renders the copper obviously unsuitable for weighing. It darkens, or forms a greyish coating on, the copper; and this darkening is a delicate test for bismuth. In assaying ores containing about three and a half per cent. of copper, and known to contain bismuth in quantities scarcely detectable in ordinary analysis, the metal deposited was distinctly greyish in colour, and would not be mistaken for pure copper. Ten grams of this impure copper were collected and analysed, with the following results:—
| Copper | 99.46 | per cent. |
| Bismuth | 00.30 | " |
| Iron | 00.14 | " |
| Arsenic | 00.10 | " |
| ——— | ||
| 100.00 |
The quantity of copper got in each assay was 0.175 gram, and consequently the bismuth averaged 0.00053 gram.
To separate the bismuth in such a case the deposit is dissolved off by warming it in the original solution. The bismuth is precipitated by the addition of ammonic carbonate, and the solution, after filtering and acidifying with nitric acid, is re-electrolysed.
Determination of Copper in Commercial Copper.—Take from 1 to 1.5 gram, weigh carefully, and transfer to a beaker; add 20 c.c. of water and 10 c.c. of nitric acid; cover with a clock[Pg 194] glass, and allow to dissolve with moderate action; boil off nitrous fumes, dilute to 100 c.c., and electrolyse. The cylinder must be carefully weighed, and the electrolysis allowed to proceed for 24 hours. The weight found will be that of the copper and silver. The silver in it must be determined[53] and deducted.
Determination of Copper in Brass, German Silver, or Bronze.—Treat in the same manner as commercial copper. If nickel is present, the few milligrams of copper remaining in the electrolysed solution should be separated with sulphuretted hydrogen, the precipitated sulphide dissolved in nitric acid, and determined colorimetrically.
There are two of these in use, one based on the decolorising effect of potassic cyanide upon an ammoniacal copper solution, and the other upon the measurement of the quantity of iodine liberated from potassic iodide by the copper salt. The cyanide process is the more generally used, and when carefully worked, "on certain understood and orthodox conditions," yields good results; but probably there is no method of assaying where a slight deviation from these conditions so surely leads to error. An operator has no difficulty in getting concordant results with duplicate assays; yet different assayers, working, without bias, on the same material, get results uniformly higher or lower; a difference evidently due to variations in the mode of working. Where a large number of results are wanted quickly it is a very convenient method. The iodide process is very satisfactory when worked under the proper conditions.
The process is based upon the facts—(1) that when ammonia is added in excess to a solution containing cupric salts, ammoniacal copper compounds are formed which give to the solution a deep blue colour; and (2) that when potassic cyanide is added in sufficient quantity to such a solution the colour is removed, double cyanides of copper and potassium or ammonium being formed.[54] In the explanation generally given the formation of cuprous cyanide is supposed[55]; but in practice it is found that one part of copper requires rather more than four parts of cyanide, which agrees with the former, rather than the latter, explanation.
Reliance on the accuracy of the process cannot rest upon the supposition that the cyanide required for decoloration is proportional to the copper present, for varying quantities of ammonia salts, ammonia and water, and differences of temperature have an important effect. The results are concordant and exact only when the cyanide is standardised under the same conditions as it is used. It is best to have the assay solution and that used for standardising as nearly as possible alike, and to titrate the two solutions side by side. This demands an approximate knowledge of the quantity of copper contained in the ore and a separation of the bulk of the impurities.
For the titration there is required a standard solution of potassium cyanide made by dissolving 42 grams of the salt, known to dealers as Potassium Cyanide (Gold), in water and diluting to one litre: 100 c.c. of this will be about equivalent to one gram of copper. For poor ores the solution may conveniently be made half this strength.
The solution of the ore and the separation of the copper as sulphide are effected in the same ways as have been already described for electrolysis. Similarly, too, the sulphide is attacked with 15 c.c. of nitric acid and the assay boiled down to 10 c.c. Add 20 c.c. of water and warm, filter into a pint flask, wash well with water, and dilute to about 150 c.c.; add 30 c.c. of dilute ammonia, and cool.
Prepare a standard by dissolving a quantity of electrotype copper (judged to be about the same as that contained in the assay) in 20 c.c. of water and 10 c.c. of nitric acid, boil off the nitrous fumes, and dilute to 150 c.c.: add 30 c.c. of dilute ammonia and cool.
Fill a burette with the standard cyanide solution. The burette with syphon arrangement, figured on page 52, is used. A number of titrations can be carried on at the same time provided the quantity of copper present in each is about the same. This is regulated in weighing up the ore. The flasks must of course be marked, and should be arranged in series on a bench in front of a good light and at such a height that the liquid can be looked through without stooping. Supposing about 50 c.c. of cyanide will be required, 30 c.c. should be run into each, and each addition be recorded as soon as made; then run 15 c.c. into each. The solutions will now probably show marked differences of tint: add 1 c.c. of cyanide to the lighter ones and more to the darker, so as to bring the colours to about the same depth of tint. They should all be of nearly equal tint just before finishing. At the end add half a c.c. at a time until the colours are completely discharged. A piece of damp filter paper held between the light[Pg 196] and the flask assists in judging the colour when nearly finished. Overdone assays show a straw yellow colour which deepens on standing.
The following will illustrate the notes recorded of five such assays and one standard:—
| (1) | 30 | c.c. | 15 | c.c. | 5 | c.c. | 2 | c.c. | 1 | c.c. | 1/2 | c.c. | — | c.c. | = | 53-1/2 | c.c. |
| (2) | 30 | " | 15 | " | 1 | " | 1 | " | 1 | " | 1/2 | " | — | " | = | 48-1/2 | " |
| (3) | 30 | " | 15 | " | 3 | " | 1 | " | 1 | " | 1/2 | " | — | " | = | 50-1/2 | " |
| (4) | 30 | " | 15 | " | 5 | " | 2 | " | 1 | " | 1/2 | " | 1/2 | " | = | 54 | " |
| (5) | 30 | " | 15 | " | 2 | " | 1 | " | 1 | " | 1/2 | " | — | " | = | 49-1/2 | " |
| (6) | 30 | " | 15 | " | 2 | " | 1 | " | 1 | " | 1/2 | " | 1/2 | " | = | 50 | standard |
Three grams of ore were taken, and the standard contained 0.480 gram of copper.
In this series the difference of half a c.c. means about 0.15 per cent. on the ore; with a little practice it is easy to estimate whether the whole or half of the last addition should be counted.
To get satisfactory results, the manner of finishing once adopted must be adhered to.
The following experiments show the effect of variation in the conditions of the assay:—Use a solution of copper nitrate, made by dissolving 10 grams of copper in 50 c.c. of water and 35 c.c. of nitric acid, and diluting to a litre. 100 c.c. = 1 gram of copper.
Effect of Varying Temperature.—In these experiments 20 c.c. of copper nitrate were used, with 10 c.c. of nitric acid, 30 c.c. of dilute ammonia, and water to 200 c.c. The results were—
| Temperature | 15° | 30° | 70° | 100° |
| Cyanide required | 21.5 c.c. | 20.8 c.c. | 19.7 c.c. | 18.8 c.c. |
The temperature is that of the solution before titrating. These show the importance of always cooling before titrating, and of titrating the assay and standard at the same temperature.
Effect of Varying Bulk.—The quantities of copper, acid, and ammonia were the same as in the last-mentioned experiments. The results were:—
| Bulk | 100.0 | c.c. | 200.0 | c.c. | 300.0 | c.c. | 400.0 | c.c. |
| Cyanide required | 23.3 | " | 21.7 | " | 21.4 | " | 21.4 | " |
These show that large variations in bulk must be avoided.
Effect of Varying Ammonia.—The quantities of copper and acid were the same as in the series of experiments last noticed. The bulk was 200 c.c. The results were:—
| Dilute ammonia | 20.0 | c.c. | 30.0 | c.c. | 50.0 | c.c. | 100.0 | c.c. |
| Cyanide required | 20.9 | " | 21.7 | " | 22.3 | " | 24.6 | " |
Effect of Varying Acid.—The quantities of copper and water were the same as in the last-noticed set of experiments: 30 c.c. of dilute ammonia were used.
| Nitric acid | 5.0 | c.c. | 10.0 | c.c. | 15.0 | c.c. |
| Cyanide required | 21.6 | " | 21.7 | " | 21.5 | " |
On adding nitric acid to the solution it combines with a portion of the ammonia to form ammonic nitrate; it will be seen from the last series of experiments that the lessening of the amount of free ammonia will decrease the quantity of cyanide required; but, on the other hand, the ammonic nitrate which is at the same time formed will increase the amount required; under the conditions of the assay these two effects neutralise each other, and such differences in the quantity of acid as are likely to occur are unimportant.
Effect of Varying Ammonic Salts.—The quantities of copper, water, and ammonia were the same as in the last mentioned set of experiments, but no nitric acid was used.
| Ammonic nitrate added | 1 gram | 5 grams | 10 grams | 20 grams |
| Cyanide required | 21.2 c.c. | 22.1 c.c. | 23.1 c.c. | 24.1 c.c. |
These show that combined ammonia seriously affects the titration, and that the principle sometimes recommended of neutralising the acid with ammonia, and then adding a constant quantity of ammonia, is not a good one, because there is then an interference both by the ammonia and by the variable quantity of ammonic salts.
The same quantity of combined ammonia has the same effect, whether it is present as sulphate, nitrate, chloride, or acetate, as the following experiments show. Four lots of 20 c.c. of "copper nitrate" were taken, and 20 c.c. of dilute ammonia added to each. These were carefully neutralised with the respective acids, rendered alkaline with 30 c.c. more of ammonia, cooled, diluted to bulk, and titrated. The results were:—
| With | sulphuric acid | 22.5 | c.c. of | cyanide |
| " | nitric acid | 22.6 | " | " |
| " | hydrochloric acid | 22.6 | " | " |
| " | acetic acid | 22.5 | " | " |
Effect of Foreign Salts.—Sulphates, nitrates and chlorides of sodium or potassium have no action, whilst the hydrates, carbonates, bicarbonates, sulphites, and nitrites have an important effect. The interference of ammonic salts has already been shown.
Salts of silver, zinc, and nickel react with cyanide just as copper does, and consequently interfere. Ferrous salts are sure to be absent, and ferric salts yield ferric hydrate with the ammonia, which is not acted on by the cyanide, but, owing to its bulkiness, it settles slowly; this lengthens the time required for titration, and so modifies the manner of working. An assay should not be worked with ferric hydrate present, unless the standard contains about the same amount of it. On mines it is often inconvenient to separate the copper by means of sulphuretted hydrogen; hence it is customary to titrate without previous[Pg 198] separation. In this case, instead of standardising the cyanide with electrotype copper, a standard ore should be used. This should be an ore (of the same kind as those being assayed) in which the copper has been carefully determined.
Effect of Varying Copper.—In these experiments 10 c.c. of nitric acid, 30 c.c. of ammonia, and water to 200 c.c. were used.
| Copper nitrate present | 1.0 | c.c. | 10.0 | c.c. | 20.0 | c.c. | 50.0 | c.c. | 100.0 | c.c. |
| Cyanide required | 0.7 | " | 11.2 | " | 21.7 | " | 54.5 | " | 108.1 | " |
These results show that under the conditions laid down the various causes of disturbance nearly neutralise one another, and the results within a fair range are practically proportional.
Determination of Copper in Copper Pyrites.—Weigh up 2 grams of the dried and powdered ore, and place in an evaporating dish about four inches in diameter. Cover with 20 c.c. of nitric acid and put on a hot plate. Evaporate to dryness without further handling. Allow to cool and take up with 30 c.c. of hydrochloric acid, boil, dilute, and transfer to a pint flask, filtering if necessary. Make up the bulk with the washings to about 150 c.c. Precipitate with sulphuretted hydrogen, filter, and wash back the precipitate into the flask. Add 15 c.c. of nitric acid, and boil down rapidly to 10 c.c. Dilute, add 30 c.c. of dilute ammonia, make up to 150 c.c., and cool. For the standard, weigh up 0.5 gram of copper, more or less, according to the quantity judged to be present in the assay. Dissolve in 20 c.c. of dilute nitric acid, boil off nitrous fumes, add 30 c.c. of dilute ammonia, make up to the same bulk as that of the assay, and cool. Titrate the two solutions side by side and as nearly as possible in the same manner.
Since the assay solution is often turbid from the presence of small quantities of lead and of iron from incomplete washing, and since this slight precipitate is very slow in settling, the standard can hardly be compared strictly with the assay. This can be counteracted by precipitating in both solutions a mixture of ferric and aluminic hydrates, which settles readily and leaves the supernatant liquor clear. To effect this, boil the nitric acid solutions with 30 c.c. of a solution containing 15 grams each of alum and ferrous sulphate to the litre. In an actual determination 2 grams of the ore were taken and compared with 0.5 gram of copper. The assay required 57.7 c.c. of cyanide and the standard 52.5 c.c.
52.5 : 0.5 :: 57.7 : 0.5495
This on 2 grams of ore = 27.47 per cent.; the same sample by electrolysis gave 27.60 per cent. of copper.[Pg 199]
Determination without Previous Separation.—Dissolve up 2 grams as before, but, instead of passing sulphuretted hydrogen, add 30 c.c. of dilute ammonia, shake well, and cool. Prepare a standard by dissolving 0.5 gram of copper in 1 c.c. of nitric acid, add 0.6 gram of iron in the form of ferric chloride and 20 c.c. of hydrochloric acid, dilute to about 150 c.c., add 30 c.c. of dilute ammonia, and cool. Titrate the two solutions side by side. In a determination on the sample last used, 58 c.c. were required for the assay and 53 c.c. for the standard, which indicates 27.3 per cent. of copper.
This method of working is somewhat rough.
This is based upon the fact that when potassic iodide in excess is added to a strong solution of a cupric salt in a faintly acid solution, cuprous iodide is formed and an equivalent of iodine liberated.[56] The iodine is measured by titrating with a solution of sodium hyposulphite,[57] using starch paste as indicator. The iodine is soluble in the excess of potassium iodide, forming a deep brown solution; the hyposulphite is added until this brown colour is almost removed. Starch paste is then added, and strikes with the remaining iodine a dirty blue colour. The addition of the "hypo" is continued until the blue colour is discharged. The end reaction is sharp; a drop is sufficient to complete it.
As regards the titration, the process leaves little to be desired; the quantity of "hypo" required is strictly proportional to the copper present, and ordinary variations in the conditions of working are without effect. The presence of salts of bismuth masks the end reaction because of the strong colour imparted to the solution by the iodide of bismuth. Under certain conditions there is a return of the blue colour in the assay solution after the finishing point has apparently been reached, which is a heavy tax on the patience and confidence of the operator. This is specially apt to occur when sodium acetate is present, although it may also be due to excessive dilution.
The standard "hypo" solution is made by dissolving 39.18 grams of the crystallised salt (Na2S2O3.5H2O) in water and diluting to one litre. One hundred c.c. will equal one gram of copper.
The starch solution is made by mixing 1 gram of starch into a thin paste with cold water, pouring it into 200 c.c. of boiling[Pg 200] water, and continuing the boiling for a minute or so. The solution must be cold before use, and about 2 c.c. is used for each assay. It should not be added until the bulk of the iodine has been reduced.
To standardise the "hypo," weigh up 0.3 or 0.4 gram of pure copper, dissolve in 5 c.c. of dilute nitric acid, boil off nitrous fumes, and dilute with an equal bulk of cold water. Add "soda" solution until a permanent precipitate is obtained, and then 1 c.c. of acetic acid. This should yield a clear solution. Fill an ordinary burette with the "hypo." Add 3 grams of potassium iodide crystals to the copper solution, and, when these are dissolved, dilute to 100 c.c. with water. Run in the "hypo" solution rather quickly until the brown colour is nearly discharged—i.e., to within 3 or 4 c.c. of the finish. Add 2 c.c. of the starch solution, and continue the addition of the "hypo" a few drops at a time until the tint suddenly changes to a cream colour. The blue colour must not return on standing three or four minutes. Calculate the standard in the usual way.
In assaying ores, the copper is dissolved and separated with sulphuretted hydrogen as in the other processes, but the sulphide should be washed more completely to ensure the absence of iron salts.
The following experiments show the effect of variation in the conditions of the assay. Use a solution of copper sulphate containing 39.38 grams of copper sulphate crystals (CuSO4.5H2O) in the litre. 100 c.c. equal 1.00 gram of copper.
Effect of Varying Temperature.—The assay after the addition of the potassic iodide must be kept cold, else iodine may be volatilised.
Effect of Varying Potassium Iodide.—In various descriptions of the process the amount of iodide required is variously stated at from "a few crystals" to as much as 10 grams. The proportion required by theory for 1 gram of copper is a little over 5 grams: an excess, however, is required to keep the liberated iodine in solution. On economic grounds this excess should not be extravagant; if the student uses 10 parts of the iodide for each part of copper in the assay he will have sufficient. In the experiments there were used 20 c.c. of the copper sulphate, with varying amounts of potassic iodide, and the following results were got:—
| Potassic iodide added | 1.5 gram | 3 grams | 5 grams |
| "Hypo" required | 20.0 c.c. | 20.0 c.c. | 20.0 c.c. |
In these the iodide was added direct to the solution containing the copper, which was afterwards diluted to 100 c.c. and titrated.[Pg 201] In another series the iodide was added after the dilution to 100 c.c., and the results were:—
| Potassic iodide added | 1.5 gram | 3 grams | 5 grams | 10 grams |
| "Hypo" required | 20.0 c.c. | 20.1 c.c. | 20.0 c.c. | 20.0 c.c. |
Effect of Varying Bulk.—In these experiments, 20 c.c. of copper sulphate were taken, 3 grams of potassic iodide added, and also water to the required bulk.
| Bulk | 20.0 | c.c. | 100.0 | c.c. | 200.0 | c.c. | 500.0 | c.c. |
| "Hypo" required | 20.0 | " | 20.0 | " | 20.0 | " | 19.9 | " |
In the last of these experiments the colour was discharged at 18 c.c., but gradually returned until 19.9 c.c. had been run in. It will be seen that considerable variation in bulk does not interfere.
Effect of Acetic Acid.—These experiments were like the last series mentioned, but the bulk was 100 c.c., and varying amounts of acetic acid were added.
| Acetic acid added | 0 | c.c. | 1.0 | c.c. | 5.0 | c.c. | 10.0 | c.c. | 20.0 | c.c. |
| "Hypo" required | 20.0 | " | 20.1 | " | 20.1 | " | 20.0 | " | 20.2 | " |
Acetic acid, then, does not interfere to any serious extent.
Effect of Varying Sodium Acetate.—These experiments were like those last mentioned, but without acetic acid, and with varying amounts of sodium acetate.
| Sodium acetate added | 0 gram | 1 gram | 2 grams | 5 grams | 10 grams |
| "Hypo" required | 20.0 c.c. | 20.0 c.c. | 20.2 c.c. | 19.3 c.c. | 18.2 c.c. |
In the 5 grams experiment, when the finishing point had been apparently reached the colour slowly returned; but as the results generally on titrating were not satisfactory a repetition of the experiment was made with the addition of 5 c.c. of acetic acid, which gave an equally bad result.
Effect of Foreign Salts.—The conditions of these experiments were the same as before. The salts were added and dissolved before the addition of the potassium iodide. Using 5 grams (or in the case of the acids, 5 c.c.), the results were as follows:—
| Salt added | — | dilute H2SO4 | Acetic acid | NaAc | NaCl |
| "Hypo" required | 20.0 c.c. | 20.0 c.c. | 20.1 c.c. | 19.3 c.c. | 20.1 c.c. |
| Salt added | KNO3 | Na2SO4 | AmCl | Am2SO4 | |
| "Hypo" required | 20.2 c.c. | 18.7 c.c. | 20.0 c.c. | 19.9 c.c. |
The low result with the sulphate of soda was evidently due to the formation of a sparingly soluble double salt, which removed copper from the solution; on adding a little acetic acid the full amount of "hypo" was required. The effect of the presence of certain metals is important. The method of determining it was to add the substance to the solution containing the copper, and[Pg 202] partly precipitate with soda solution; then treating with 1 c.c. of acetic acid, adding the iodide, and proceeding as before.
| Substance Added. | "Hypo" Required, |
| - | 20. c.c |
| 0.050 gram arsenic as As2O5 | 20.0 " |
| 0.050 " antimony as SbCl5 | 19.8 " |
| 0.050 " lead as Pb(NO3)2 | 20.1 " |
A similar experiment with 0.050 gram of bismuth nitrate could not be determined, the end-reaction being masked. Bismuth iodide is soluble in potassic iodide, forming a brown solution, the colour of which is very similar to that produced by iodine; and although it does not strike a blue colour with starch, "hypo" has an action on it.
A similar experiment with 0.050 gram of iron as ferric chloride required 22.3 c.c. of "hypo," and the colour returned on standing. This shows that ferric acetate liberates iodine under the conditions of the assay. Trying to counteract this, by adding to a similar solution 0.5 gram of phosphate of soda dissolved in a little water, 19.7 c.c. of "hypo" were required instead of 20.0, but the assay showed signs of returning colour.
In standardising, the same result was obtained, whether the copper was present as nitrate or sulphate before neutralising.
Effect of Varying Copper.—With the same conditions as before, but with varying amounts of copper and a proportionally increasing quantity of iodide, the results were:—
| Copper present | 1.0 | c.c. | 10.0 | c.c. | 20.0 | c.c. | 50.0 | c.c. | 100.0 | c.c. |
| "Hypo" required | 1.0 | " | 10.0 | " | 20.0 | " | 50.0 | " | 100.0 | " |
showing the results to be exactly proportional.[58]
Determination of Copper in Copper Pyrites.—Take 2 grams of the dried and powdered ore and treat in a porcelain dish with 20 c.c. of nitric acid, and evaporate to dryness. Take up with 30 c.c. of hydrochloric acid, dilute, and transfer to a pint flask; make up with water to 200 c.c., warm, and pass sulphuretted hydrogen to excess. Filter, and wash the precipitate with water acidified with sulphuric acid. Wash the precipitate back into the flask, and dissolve with 15 c.c. of nitric acid. Evaporate almost to dryness; add 20 c.c. of water, and boil till free from nitrous fumes; filter off the sulphur and gangue; neutralise with soda, avoiding excess; add 1 or 2 c.c. of acetic acid, and shake till clear. Add 5 grams of potassium iodide, dilute to 100 c.c., and titrate. The following is an example:—
| 0.5 gram of copper required | 50.5 | c.c. | "hypo." |
| The assay required | 55.6 | " | " |
which is equal to 27.5 per cent. of copper.[Pg 203]
This is based on the blue coloration of ammoniacal copper solutions. The quantity of copper in 100 c.c. of the assay solution should not be more than 15 milligrams, or less than half a milligram. It is not so delicate as most other colorimetric methods, but nevertheless is a very useful one.
The manner of working is the same as that described under iron.
Standard Copper Solution.—Weigh up 0.5 gram of electrotype copper, dissolve in 10 c.c. of nitric acid, boil off nitrous fumes, and dilute to 1 litre. 1 c.c. = 0.5 milligram.
In nearly all cases it will be necessary to separate the copper with sulphuretted hydrogen from a solution of about 5 grams of the material to be assayed. The filter paper containing the sulphide (and, probably, much sulphur) is dried and burnt. The ashes are dissolved in 5 c.c. of dilute nitric acid, 10 c.c. of dilute ammonia added, and the solution filtered through a coarse filter into a Nessler tube, washing the paper with a little dilute ammonia.
The estimation of the colour and calculation of the result are made in the way described on page 44.
The effect of varying conditions on the assay may be seen from the following experiments.
Effect of Varying Temperature.—The effect of increased temperature is to slightly decrease the colour, but this can only be observed when a fair quantity of copper is present.
| 1.0 | c.c. at 15° | showed the | colour of | 1.0 | c.c. at 70° |
| 2.5 | " | " | " | 2.7 | " |
| 5.0 | " | " | " | 5.0 | " |
| 10.0 | " | " | " | 9.0 | " |
Effect of Varying Ammonia.—The solution must, of course, contain free ammonia; about 5 c.c. of dilute ammonia in 50 c.c. bulk is the quantity to be used in the experiments. A larger quantity affects the results, giving lower readings and altering the tint. With small quantities of ammonia the colour approaches a violet; with larger, a sky-blue.
| 2.5 | c.c. with | 25 | c.c. of | strong ammonia | read | 2.2 | c.c. |
| 5.0 | " | " | " | " | " | 4.0 | " |
| 10.0 | " | " | " | " | " | 8.0 | " |
Effect of Ammonic Salts.—The following table shows the results after addition of ammonic salts:[Pg 204]—
| C.c. Present. | With 10 grams Ammonic Nitrate. | With 10 grams Ammonic Chloride. | With 10 grams Ammonic Sulphate. |
| 2.5 | 2.5 | 2.5 | 2.0 |
| 5.0 | 5.0 | 5.3 | 4.3 |
| 10.0 | 10.0 | 10.0 | 8.5 |
These show that sulphates should be avoided, and either nitrate or chloride solutions be used in the standard as well as in the assay.
Determination of Copper in a Manganese Ore.—Treat 3 grams of the ore with 20 c.c. of hydrochloric acid, and evaporate to dryness. Take up with 10 c.c. of hydrochloric acid; dilute to about 200 c.c., and pass sulphuretted hydrogen until the solution smells of the gas; filter, burn, take up with 5 c.c. of dilute nitric acid, add 10 c.c. of dilute ammonia, and filter into the Nessler tube, and make up with the washings to 50 c.c. Into the "standard" tube put 5 c.c. of dilute nitric acid and 10 c.c. of dilute ammonia. Make up to nearly 50 c.c. with water, and run in the standard copper until the colours are equal. In a determination 4 c.c. (= 2.0 milligrams of copper) were required; this in 3 grams of ore = 0.07 per cent.
Determination of Copper in "Black Tin."—Weigh up 3 grams of the dried ore, boil with 10 c.c. of hydrochloric acid, and afterwards add 1 c.c. of nitric; boil off nitrous fumes, evaporate to about 5 c.c., dilute to 50 c.c., add 20 c.c. of dilute ammonia; stir, and filter. If much iron is present, dissolve the precipitate of ferric hydrate in acid, and reprecipitate with ammonia. Mix the two filtrates, and dilute to 100 c.c. Take 50 c.c. for the test. A sample of 3 grams of an ore treated in this way required 5.2 c.c. of standard copper to produce equality of tint. This gives 0.35 per cent.
Determination of Copper in Tin.—Weigh up 1 gram of the sample, transfer to an evaporating dish, and cover with 30 c.c. of aqua regia. Warm until the metal has dissolved, then evaporate almost to dryness. Take up with a few c.c. of hydrochloric acid and again evaporate.
Dissolve the residue in 10 c.c. of dilute hydrochloric acid and transfer to a 100 c.c. flask. Add 10 c.c. of dilute ammonia and make up with water to the containing mark.
Filter off 50 c.c. of the solution into a Nessler glass and determine the copper in it colorimetrically.[Pg 205]
Very pure copper can be obtained in commerce, owing to the demand for metal of "high conductivity" for electrical purposes, which practically means for metal free from impurities.
Much of the metal sold contains as much as one per cent. of foreign substances, of which arsenic is the most important. The other elements to be looked for are bismuth, lead, antimony, silver, gold, iron, nickel, cobalt, sulphur, and oxygen. In "blister copper" (which is the unrefined metal), aluminium, silicon, and phosphorus may be met with.
Oxygen.—All commercial copper carries oxygen; most of it is present as cuprous oxide, which is dissolved by molten copper. The estimation of oxygen is often made "by difference." The copper and the other impurities being determined, the rest is assumed to be oxygen. Probably this is nearly correct, but the whole of the oxygen should not be ascribed to cuprous oxide; for any arsenic the metal contained would be present as cuprous arsenite, since arsenide of copper and cuprous oxide could not exist together at the temperature of fusion without interacting. In the report of the analysis, it is best to state the proportion of oxygen thus:—
Oxygen ——— per cent. by difference.
There is a method of determination by fusing 5 or 10 grams in a brasqued crucible, and counting the loss as oxygen; and another method for the determination of cuprous oxide based on the reaction of this substance with nitrate of silver.[59] About 2 grams of silver nitrate, dissolved in 100 c.c. of water, is allowed to act upon 1 gram of the copper in the cold. The precipitate is filtered off, washed thoroughly with water, and the basic salt dissolved and determined colorimetrically.
One part of copper found represents 1.68 part of cuprous oxide, or 0.19 part of oxygen. Copper generally carries from 0.1 to 0.2 per cent. of oxygen.
Silver is found in most samples, but occurs in variable proportions; when it amounts to 30 ounces per ton it has a commercial value. To determine its amount, dissolve 10 grams of the copper in 35 c.c. of nitric acid and 50 c.c. of water, boil off nitrous fumes, and dilute to about 100 c.c. One or two c.c. of dilute hydrochloric[Pg 206] acid (one to 100 of water) are added, stirred in, and the precipitate allowed to settle for twenty-four hours. Filter through a double Swedish paper, dry, burn, and cupel the ashes with one gram of sheet lead.
Ten grams of a sample of copper gave in this way 4.7 milligrams of silver. Ten grams of the same copper, to which 24 milligrams of silver had been added gave 28.2 milligrams.
Gold.—To determine it, dissolve 10, 20, or 50 grams of the sample in 35, 70, or 175 c.c. of nitric acid and an equal volume of water, boil till free from nitrous fumes, and dilute to double its volume. Allow to stand for some time, decant on to a filter, dry, burn, and cupel the ashes with 1 gram of sheet lead. If silver is present, owing to traces of chlorides in the re-agents used, "parting" will be necessary. (See Gold.)
Working in this way on 20 grams of copper, to which 1.8 milligram of gold had been added, a button weighing 2.0 milligrams was obtained.
Antimony is not a frequent impurity of copper: it can be detected in quantities over 0.1 per cent. by a white residue of Sb2O4, insoluble in nitric acid. With material containing only small quantities of antimony the white oxide does not show itself for some time, but on long-continued boiling it separates as a fine powder. It is best (when looking for it) to evaporate the nitric acid solution to the crystallising point, to add a little fresh nitric acid and water, and then to filter off the precipitate. After weighing it should be examined for arsenic and bismuth.
Lead.—Refined coppers are often free from lead, anything more than traces being seldom found; in coarse coppers it is sometimes present in considerable quantities.
Its presence may be detected in the estimation of the copper electrolytically, the platinum spiral becoming coated with a brown or black deposit of lead dioxide. The depth of colour varies with the lead present, and obviously could be made the basis of an approximate estimation. The colour shows itself within an hour or so, but is best observed when all the copper has been deposited.
Electrolysing a solution of one gram of pure copper, to which 0.5 milligram of lead had been added, the deposit was dark brown; in a similar solution with 1 milligram of lead it was much darker, and with 2 milligrams it was black. Under the conditions of the assay the dioxide cannot be weighed, as it partly dissolves on breaking the current. When lead has been found, its quantity may be estimated by evaporating to dryness the nitric acid solution to which an excess of sulphuric acid has been added, taking up with water, and filtering off and weighing the lead sulphate.[Pg 207]
The separation of traces of lead as chromate is a fairly good one. Dissolve 5 grams of the copper in 17 c.c. of nitric acid and an equal volume of water; boil off nitrous fumes, neutralise with soda, and afterwards acidulate with acetic acid; and dilute to a litre. Add 20 grams of sodium acetate, warm, and precipitate the lead with a dilute solution of potassium chromate. Copper chromate (yellow) may be at the same time thrown down, but it is readily soluble on diluting. Filter off the precipitate; wash it into a beaker and pass sulphuretted hydrogen; oxidise the sulphide and weigh as lead sulphate. Treated in this way 5 grams of copper yielded sulphate of lead equal to 2.0 milligrams of lead. Five grams of the same sample to which 10 milligrams of lead were added gave 11.4 milligrams.
Nickel and Cobalt.—Nickel is always present in larger or smaller quantities in commercial copper, and, perhaps, has an influence on the properties of the metal. It is determined as follows:—Dissolve 10 grams of the copper in 35 c.c. of nitric acid and an equal bulk of water, boil off nitrous fumes and neutralise with soda, add 2 grams of carbonate of soda dissolved in water, boil, and filter. Acidify the filtrate with 2 or 3 c.c. of dilute nitric acid and dilute to 1 or 1-1/2 litres. Pass sulphuretted hydrogen through the cold solution till the copper is all down and the liquid smells of the gas. Filter and evaporate the filtrate to a small bulk, and determine the nickel by electrolysing the solution rendered ammoniacal, or by precipitating as sulphide and weighing as sulphate. (See under Nickel.) The precipitate, after weighing, should be tested for cobalt. If present it is separated with potassium nitrite as described under Cobalt. Ten grams of copper gave 6.0 milligrams of nickel; and another lot of 10 grams of the same copper, to which 10.0 milligrams of nickel had been added, gave 17.2 milligrams.
Sulphur.—The amount of sulphur in refined copper is very small, seldom exceeding 0.005 per cent. In coarse copper, as might be expected, it is found in larger quantities.
In determining it, it is first converted into sulphuric acid, and then precipitated and weighed as barium sulphate. The precipitation cannot be effected from a nitric acid solution. Ten grams of copper are dissolved in nitric acid, as for the other determinations, and then boiled with excess of hydrochloric acid till the nitric acid is completely removed. There is then added a few drops of a dilute solution of baric chloride, and the solution is allowed to stand for some hours. The baric sulphate is filtered off and weighed.
The necessity for precipitating from a hydrochloric acid solution is seen from the following determinations. In each experiment[Pg 208] 10 grams of copper was used, and a known weight of sulphur, in the form of copper sulphate, added.
| Sulphur added. | Sulphur found in Hydrochloric Acid Solution. | Sulphur found in Nitric Acid Solution. |
| 5 milligrams | 8 milligrams | 0.03 milligrams |
| 10 " | 11 " | 0.03 " |
| 15 " | 17 " | 12.00 " |
Bismuth.—Nearly all samples of copper contain bismuth, but only in small quantities. It is best determined colorimetrically as described under Bismuth. The method of concentrating and preparing the solution for colorimetric assay is as follows. Dissolve 10 grams of copper in nitric acid, as in the other determinations; neutralise with soda; add 1 or 1.5 grams of bicarbonate of soda and boil for ten minutes; filter, dissolve the precipitate in hot dilute sulphuric acid; add sulphurous acid and potassium iodide in excess, and boil till free from iodine. Filter and dilute to 500 c.c. Take 50 c.c. of the yellow solution for the determination. A few c.c. of a dilute solution of sulphurous acid (1 in 100) will prevent the liberation of iodine. The following experiments test the method of separation. Ten grams of copper were treated as above and precipitated with 1.5 gram of "soda;" the precipitate contained 0.6 milligram of bismuth (= 0.006 per cent.). The filtrate treated with another 1.5 gram of "soda" gave a precipitate which was free from bismuth. To the filtrate from this was added 1.0 milligram of bismuth, and another fraction was precipitated with 1.5 gram of "soda." In this precipitate was found 1.0 milligram of bismuth. To the filtrate another milligram of bismuth was added and the separation with "soda" repeated. The bismuth was separated from this precipitate with ammonic carbonate before determination, and 0.9 milligram was found.
Arsenic.—The proportion of arsenic in copper varies from 0.01 to 0.75 per cent. whilst in coarse copper it may amount to 2 or even 3 per cent. To determine it, dissolve 5, 10, or 20 grams of the copper (according to the amount of arsenic present) in 18 c.c., 35 c.c., or 70 c.c. of nitric acid, and an equal volume of water. Boil off the nitrous fumes, dilute to 100 c.c. and neutralise with soda; add 1.5 or 2 grams of carbonate of soda dissolved in a little water, and boil. Filter (washing is unnecessary) and dissolve back into the flask with a little dilute hydrochloric acid; add 30 c.c. of dilute ammonia and 25 c.c. of "magnesia mixture," and allow to stand overnight. The whole of the arsenic is precipitated as ammonic-magnesic arsenate in one hour, but it is[Pg 209] advisable to leave it longer. The precipitate may be dried and weighed, or, better, titrated with uranium acetate. (See Arsenic.) To test this method of separation 10 grams of pure copper were taken and 0.200 gram of arsenic dissolved with it. The arsenic was determined by titration with uranium acetate, and 0.200 gram was found. Two other similar experiments with 0.080 and 0.010 gram of arsenic added, gave 0.079 and 0.012 gram respectively.
Antimony or bismuth may be present without interfering with the titration. With 0.100 gram of antimony and 0.100 gram of arsenic, 0.100 gram of arsenic was found; and in another case, with 0.100 gram of bismuth and 0.060 gram of arsenic, 0.060 gram was found. In these experiments the antimony and bismuth were present in the assay solution when titrated. For a gravimetric determination they would require to be removed before precipitating with "magnesia mixture."
Phosphorus, if present, counts as arsenic in the proportion of 1 to 2.4; but, except in the case of coarse coppers, it is always absent.
Iron, if present, interferes by forming a white flocculent precipitate of ferric arsenate after the addition of the sodium acetate and preliminary to the titration. Each milligram of iron abstracts, in this way, 1.3 milligrams of arsenic.
Iron.—Refined coppers carry traces of iron, varying from 0.001 to 0.01 per cent. It is best determined during the arsenic estimation. The precipitate of the ammonic-magnesic arsenate will contain the whole of the iron as ferric hydrate. On dissolving in hydrochloric acid, neutralising with ammonia, adding 5 c.c. of sodic acetate, diluting, and boiling, it reappears as a white precipitate of ferric arsenate. It is filtered off (the arsenic being estimated in the filtrate), dissolved in warm hydrochloric acid, and determined colorimetrically as described under Iron. A series of experiments testing the separation is there given.
Phosphorus.—Refined coppers do not carry phosphorus, although it may be present in "coarse copper" up to 1 per cent. or more. In such samples the following method is adopted for the estimation of both phosphorus and arsenic. Dissolve 10 grams of copper and 0.1, 0.2, or 0.3 gram of iron wire (according to the amount of arsenic and phosphorus present) in 35 c.c. of nitric acid and an equal volume of water. Add soda till the free acid is nearly neutralised. Next add a strong solution of sodium acetate, until the solution ceases to darken on further addition, then dilute with water to half a litre. The solution is best contained in a large beaker; it is next heated to the boiling point, and at once removed and allowed to settle. If the precipitate is[Pg 210] light coloured it is evidence that sufficient iron has not been added, or, if it is green, from basic copper salts, it shows that the solution was not sufficiently acid. In either case start afresh. Filter off the precipitate and wash with hot water containing a little sodium acetate, dissolve it off the filter with hot dilute hydrochloric acid, add ammonia in excess, and pass sulphuretted hydrogen for five minutes. Warm at about 70° C. for a quarter of an hour. Filter. The clear yellow filtrate contains the arsenic and phosphorus. Add dilute sulphuric acid in excess; filter off the yellow precipitate of sulphide of arsenic, dissolve it in nitric acid, and titrate with uranium acetate, as described under Arsenic.
The filtrate from the sulphide of arsenic is rendered alkaline with ammonia and "magnesia mixture" added. The solution is stirred, and allowed to stand overnight. The precipitate of ammonic-magnesic phosphate is filtered off, dissolved, and titrated with uranium acetate, using the same standard solution as is used in the arsenic assay: 0.5 gram of arsenic equals 0.207 gram of phosphorus.
Copper.—The method of determining this has been described under Electrolytic Assay.
In the method of concentration by fractional precipitation with sodic carbonate (which is adopted in most of these determinations) the precipitate will contain all the bismuth, iron, and alumina; the arsenic and phosphorus as cupric arsenate and phosphate; and the greater part of the lead, antimony, and silver. The nickel and cobalt, and the sulphur as sulphuric acid, will remain in solution with the greater part of the copper.
1. According to a wet assay 2 grams of a certain ore contained 0.3650 gram of copper. What would you expect the dry assay produce to be?
2. A standard solution is made by dissolving 25 grams of potassic cyanide and diluting to a litre. Assuming the salt to be 98 per cent. real cyanide, what would 100 c.c. of the solution be equivalent to in grams of copper?
3. How would you make a solution of "hypo" of such strength that 100 c.c. shall equal 0.633 gram of copper?
4. What weight of ore, containing 17.0 per cent. of copper, would you take in order to get about 0.5 gram of copper in solution for electrolysis?
5. The solution of copper in nitric acid is effected by the following reaction:—
3Cu + 8HNO3 = 3Cu(NO3)2 + 4H2O + 2NO.
What volume of nitric acid will be required to dissolve 1 gram of copper?[Pg 211]
The chief ore of lead is galena, a sulphide of lead, common in most mining districts, and frequently associated with blende and copper-pyrites. It always carries more or less silver; so that in the assay of the ore a silver determination is always necessary. Carbonate (cerussite), sulphate (anglesite), and phosphate (pyromorphite) of lead also occur as ores, but in much smaller quantities.
Lead ores are easily concentrated (owing to their high specific gravity, &c.) by mechanical operations, so that the mineral matter sent to the smelter is comparatively pure.
Lead is readily soluble in dilute nitric acid. The addition of sulphuric acid to this solution throws down heavy, white, and insoluble lead sulphate.
Galena is soluble in hot hydrochloric acid, sulphuretted hydrogen being evolved; but the action is retarded by the separation of the sparingly soluble lead chloride. If a rod of zinc is placed in this solution, metallic lead is precipitated on it as a spongy mass, the lead chloride being decomposed as fast as it is formed. The opening up of the ore is thus easily effected, the sulphur going off as sulphuretted hydrogen, and the lead remaining in a form easily soluble in dilute nitric acid. Galena itself is readily attacked by nitric acid, part of the lead going into solution, and the rest remaining as insoluble lead sulphate. The sulphate is due to the oxidation of the sulphur by nitric acid; its amount will vary with the quantity and concentration of the acid used. Sulphate of lead is soluble in solutions of ammonium or sodium acetate; or it may be converted into carbonate by boiling with carbonate of soda. The carbonate, after washing off the sulphate of soda, dissolves easily in nitric acid. The precipitation of lead from acid solutions with sulphuric acid, and the solubility of the precipitate in ammonium acetate, distinguishes it from all other metals. The addition of potassium chromate to the acetate solution reprecipitates the lead as a yellow chromate.
The dry assay of lead is largely used, but it is only applicable to rich or concentrated ores, and even with these only gives approximate results. Both lead and lead sulphide are sensibly volatile at a moderately-high temperature; hence it is necessary to obtain a slag which is easily fusible. As a reducing agent iron is almost always used, and this is added either in the form of an iron rod, or the crucible itself is made of this metal. The flux used is carbonate of soda.[Pg 212]
When a clay crucible is used, the method of working is as follows:—Weigh up 25 grams of the dry and powdered ore, mix with an equal weight of "soda" and 2 grams of tartar; place in a crucible (E. Battersea round), and then insert a piece of iron rod about half an inch in diameter, and of such a length that it will just allow the crucible to be covered. The rod should be pushed down so as to touch the bottom of the crucible, and the mixture should be covered with a sprinkling of borax. Place in a furnace heated to, but not above, redness, and cover the crucible. In about twenty minutes the charge will be fused: the fusion is complete when bubbles of gas are no longer being evolved; and then, but not till then, the iron is withdrawn, any adhering buttons of lead being washed off by dipping the rod a few times in the slag. Cover the crucible, leave it for a minute or two, and then pour. Detach the slag, when cold, by hammering. The weight of the button multiplied by 4 gives the percentage. The commoner errors of students in working the process are too high a temperature and too quick a withdrawal.
A sample of ore treated in this manner gave on duplicate assay 17.5 and 17.6 grams of lead, equalling 70.0 and 70.4 per cent. respectively. By wet assay the sample gave 73.3 per cent. Using an iron crucible, the results will be 1 per cent. or so higher. The crucible must be made of wrought iron; and, if it has been previously used, should be cleaned by heating to dull redness and scraping the scale off with a stirrer. Take 30 grams of the ore, mix with 30 grams of "soda" and 3 grams of tartar; put the mixture in the crucible, and cover with a sprinkling of borax; heat for about twenty minutes at not too high a temperature, and then scrape down the slag adhering to the side with a stirrer. Leave in the furnace till action has ceased. Before pouring, tap the pot gently, and then tilt it so as to make the slag wash over the part of the crucible along which the charge is to be poured. Pour; and, when cold, clean and weigh the button of metal. A crucible may be used from ten to twenty times.
These assays are for ores containing the lead chiefly as sulphide. For oxidised ores, charcoal or tartar is employed as the reducing agent. The student may practise on red lead as follows:—Take 30 grams of red lead; mix with 10 grams each of borax and "soda" and about 1.5 gram of powdered charcoal; place in a small clay crucible with a cover (C. Battersea round), fuse at a gentle heat, and pour when action ceases. This assay will only take a few minutes.
Where lead is present as phosphate (as in the case of pyromorphite), or mixed with phosphates (as sometimes happens), carbonate of soda is a suitable flux; but the phosphate of soda[Pg 213] which is formed makes a thick tenacious slag, which is very apt to be carried out of the pot by the escaping gas. A wide-mouthed clay pot is taken and a little fluor spar added. For the assay of pyromorphite the following charge may be used:—Ore, 20 grams; "soda," 25 grams; tartar, 7 grams; and fluor spar, 5 grams; and 2 grams of borax as a cover. This will melt down in about ten minutes, and should be poured as soon as tranquil.
In the case of galena the best method of getting the lead into solution is to treat with hydrochloric acid and zinc. Put 1 gram of the ore in an evaporating dish 4 inches across, and cover with 10 c.c. of dilute hydrochloric acid. Heat till the evolution of sulphuretted hydrogen becomes sluggish, and then drop in a piece of zinc rod. If the solution effervesces too strongly, dilute it. Continue the heating until the sulphide is seen to be all dissolved; when the lead is all precipitated, pour off the liquid and wash twice with cold water. Peel off the precipitated lead with the help of a glass rod, and then clean the zinc. Cover the lead with 20 c.c. of water and 5 c.c. of dilute nitric acid, and heat gently till dissolved; all the lead will be in solution, and, when filtered off from the gangue, will be ready for a gravimetric determination. For volumetric work this filtering is unnecessary.
The chief objection to this method is that commercial zinc carries considerable quantities of lead. Although this can be determined and allowed for, the correction required is in most cases too large to be satisfactory. The following method is applicable in all cases, but is more troublesome:—Treat 1 gram of the ore with 10 c.c. of dilute nitric acid in an evaporating dish covered with a clock-glass, and evaporate till nearly dry. Take up with 50 c.c. of water, and add 10 c.c. of dilute sulphuric acid. Filter. The residue contains the lead as sulphate, together with the insoluble matter of the ore and globules of sulphur. Warm with a solution of ammonium acetate, and filter. The lead will be in the filtrate, and is recovered in a state fit for direct gravimetric estimation by the addition of dilute sulphuric acid. If the volumetric method is to be used, the lead sulphate should be dissolved out with a solution of sodium acetate instead of with the ammonium salt solution.
The lead is separated and precipitated as sulphate, as already described. The solution must be allowed to stand, and the clear[Pg 214] liquid be decanted through a filter. Transfer the precipitate, and wash with very dilute sulphuric acid (1 or 2 c.c. in 100 c.c. of water). The acid must be completely removed with one or two washes with cold water, and then with alcohol. The volume of liquid required for washing is small, as the precipitate is dense and easily cleaned; but the washing must be carefully done, since if any acid remains it will, on drying, char the paper, and render the subsequent work troublesome. Dry, transfer to a watch-glass, and burn the filter paper, collecting its ash in a weighed porcelain crucible. The filter paper must be freed as much as possible from the lead sulphate before burning, and the ash treated with a drop or two of nitric and sulphuric acids. Transfer the lead sulphate to the crucible; ignite gently, keeping the temperature below redness; cool, and weigh. The precipitate will contain 73.6 per cent. of lead oxide or 68.3 per cent. of lead.
Determination of Lead in Commercial Zinc.—Take 10 grams of zinc, and treat (without heating) with 60 c.c. of dilute hydrochloric acid. When the zinc is nearly all dissolved, decant off the clear liquid, and dissolve the residue in 2 c.c. of dilute nitric acid. Evaporate till most of the acid is removed; dilute to 20 or 30 c.c. with water, and add 10 c.c. of dilute sulphuric acid. Filter off, and weigh the lead sulphate. Ten grams treated in this way gave—0.1610 gram of lead sulphate, equivalent to 1.10 per cent. of lead.
This is based upon the reaction between chromate of potash and soluble lead salts in neutral solutions, whereby an insoluble yellow chromate of lead is produced.[60] An excess of the chromate is required to complete the reaction, so that the point at which an indicator shows the presence of undecomposed chromate cannot be satisfactorily taken as the finish. Therefore an excess of the standard chromate must be run in, and such excess determined.
Chromate of lead is not precipitated from strong nitric acid solutions, and only incompletely from dilute ones. Acids generally are detrimental to the precipitation, and must be neutralised before titrating. If the lead is present as sulphate in sodic acetate solution, it is well to render it distinctly alkaline with ammonia.
Lead chromate precipitated in the cold is a lemon-yellow, light precipitate, very difficult to filter: on heating to 40° C. the colour becomes orange; at 60° C. it assumes a deeper hue, and becomes[Pg 215] flocculent; and at a boiling temperature it still further darkens and settles readily. These changes in colour are not due to any chemical change, as will be seen by testing the filtrate for chromium or lead: this is an advantage to the assay, since it is only at the higher temperature that the precipitate can be easily filtered. The lead is not completely precipitated, but the amount remaining in solution is only 2 or 3 milligrams, which is just sufficient to give a dark coloration with sulphuretted hydrogen.
The standard chromate of potash solution is made by dissolving 7.13 grams of bichromate of potash and 2.0 grams of caustic soda in water, and diluting to 1 litre; or 9.40 grams of the neutral chromate (K2CrO4) may be dissolved and diluted to 1 litre: 100 c.c. will be equivalent to 1.000 gram of lead.
Standard Lead Solution.—16 grams of nitrate of lead (Pb(NO3)2) are dissolved in water and diluted to 1 litre; 100 c.c. will contain 1.000 gram of lead.
Acetate of Soda Solution.—250 grams of the crystallised salt (NaAc.3H2O) are dissolved, and diluted to 1 litre. Use 40 c.c. for each assay.
In the titration the assay solution should measure 150 to 200 c.c., and should be boiling or nearly so. It is best contained in a pint flask, and the standard chromate solution used with an ordinary burette. Run in the chromate solution in a steady stream until the whole of the lead has been precipitated. The amount required for this may be calculated: for example, 1 gram of an 80 per cent. ore would require 80 c.c. A little of the assay may be filtered off, and if it does not show a yellow colour in the filtrate run in 2 c.c. more of the standard solution and continue this addition till a colour is shown. After this run in another c.c. to ensure an excess, dilute to 250 c.c., and heat to boiling; allow to settle for three or four minutes, filter off 50 c.c. into a Nessler glass, and determine the excess of chromate colorimetrically. The excess found in the 50 c.c. must, of course, be multiplied by five, and then be deducted from the quantity of chromate originally run into the assay solution. The quantity to be deducted should not exceed 3 c.c. Where a number of determinations are made the colorimetric estimation is facilitated by using a series of standard phials similar to those described under the Electrolytic Copper Assay. The determination is rendered sharper and less liable to error by the addition of a few drops of acetic acid to convert the chromate into bichromate. The same chromate solution must be used in this determination as was used in the precipitation.
In standardising the chromate solution, the standard lead nitrate solution is used. A quantity containing about as much[Pg 216] lead as the assay is supposed to contain is measured off, rendered alkaline with dilute ammonia, and then neutralised with acetic acid, using a small piece of litmus paper dropped into the solution as indicator. Then dilute, boil, and titrate. When the lead in the assay has been separated as sulphate and dissolved in sodic acetate, less chromate is apparently required, and in this case it will be necessary to precipitate the lead in the standard with an equivalent of sodic sulphate and redissolve in sodic acetate just as in the assay. In these solutions (although there is considerable chromate in excess) a further addition of 5 or 6 c.c. of the chromate solution will cause a further precipitate. The following experiments show the effect of variation in the conditions of the assay:—
Effect of Varying Temperature.—Twenty c.c. of lead nitrate solution and 10 grams of sodium acetate were used; diluted to 200 c.c., heated to the desired temperature, and titrated. The results were:—
| Temperature | 15° | 30° | 50° | 100° |
| "Chromate" required | 19.8 c.c. | 19.5 c.c. | 19.3 c.c. | 19.2 c.c. |
The first two of these filtered badly, the precipitate coming through the filter; the last was very satisfactory in the working.
Effect of Varying Bulk.—Using 20 c.c. of lead nitrate, and 10 grams of sodium acetate as before, diluting to the required bulk, heating to boiling, and titrating, the results were:—
| Bulk | 100.0 | c.c. | 200.0 | c.c. | 500.0 | c.c. | 1000.0 | c.c. |
| "Chromate" required | 19.6 | " | 19.3 | " | 19.4 | " | 19.4 | " |
Effect of Varying Acetic Acid.—Since the experiments are carried out in the presence of sodic acetate, acetic acid is the only acid whose effect need be considered. Working as before, but with 200 c.c. bulk and varying amounts of the acid, the results were:—
| Acid present | — | 10.0 | c.c. | 20.0 | c.c. | 40.0 | c.c. |
| "Chromate" required | 19.7 c.c. | 19.1 | " | 18.5 | " | 17.3 | " |
These experiments show that only slight quantities of acid are admissible.
Effect of Varying Sodium Acetate.—With the same conditions as before, but with varying weights of sodium acetate, the results were:—
| Sodium acetate present | — | 5 grams | 10 grams | 25 grams | 50 grams |
| "Chromate" required | 19.7 c.c. | 19.6 c.c. | 19.6 c.c. | 18.8 c.c. | 17.8 c.c. |
These experiments show that excessive quantities of sodium acetate must be avoided. Ammonium acetate interferes to a[Pg 217] greater extent, and if both acetic acid and this salt are present, each exerts its disturbing influence. With 10 grams of ammonium acetate, 19.4 c.c. of the chromate solution were required instead of 19.7 c.c. in the absence of this salt; with 10 grams of the acetate and 10 c.c. of acetic acid, only 18.6 c.c. were required.
Effect of Foreign Salts.—As already stated, sulphates interfere. Twenty c.c. of the lead nitrate solution were taken, precipitated with sulphate of soda, and the precipitate dissolved in 10 grams of sodium acetate and titrated as before. Duplicate experiments required 18.6 c.c. and 18.7 c.c. of the chromate solution. A similar experiment with 40 c.c. of lead nitrate required 37.4 c.c. of chromate. If the sulphate had not been present, the results would have been 19.7 c.c. and 39.4 c.c. respectively.
Effect of Varying Lead.—In these experiments the conditions were as before, but with varying amounts of lead.
| Lead nitrate solution present | 10.0 | c.c. | 20.0 | c.c. | 50.0 | c.c. | 100.0 | c.c. |
| Chromate solution required. | 9.4 | " | 19.7 | " | 48.8 | " | 98.2 | " |
Determination of Lead in Galena.—Weigh up 1 gram of the powdered and dried ore, and boil in an evaporating dish with 10 c.c. of dilute hydrochloric acid. When the action becomes sluggish, dilute with an equal bulk of water, and add a weighed piece of zinc rod about 1 inch long and quarter-inch across. Keep up a moderate action by warming till the ore is seen to be completely attacked and the lead precipitated. Decant off the solution, wash once, strip off the lead, wash and weigh the remaining zinc. Dissolve the lead in 5 c.c. of dilute nitric acid, and 5 c.c. of water with the aid of heat. Dilute and transfer to a pint flask; add a slight excess of dilute ammonia, and render faintly acid with acetic acid. Dilute to 150 c.c., heat to boiling, and run in the standard chromate in slight excess, noting the amount required, and make up to 250 c.c. with water. Boil the solution, allow to settle for a minute or so, filter off 50 c.c., and determine the excess of chromate colorimetrically. As an example, 1 gram of an impure galena was precipitated with 75 c.c. of standard chromate (100 c.c. = 1.020 gram lead). The excess found in 50 c.c. was 0.3 c.c., which, multiplied by 5, gives 1.5 c.c. as the excess in the whole solution. The remaining 73.5 c.c. of "chromate" required by the assay, are equivalent to 0.7497 gram of lead. The zinc used up weighed 1.5 grams, and contained 0.0165 gram of lead. Thus we get—
| Lead in the assay | 0.7497 | gram |
| Lead from the zinc | 0.0165 | " |
| ——— | ||
| ∴ Lead in the galena | 0.7332 | " |
Equivalent to 73.3 per cent.
Another sample, in which the galena was accompanied with a large quantity of pyrites, gave the following results:—Three grams were treated with 30 c.c. of dilute hydrochloric acid and a rod of zinc. The zinc and lead were carefully transferred to another vessel, the zinc cleaned, and the lead (dissolved in 5 c.c. of dilute nitric acid and 20 c.c. of water) treated as before.
| 14.5 c.c. of the chromate were required | = 0.1479 | gram lead |
| Lead in 2 grams of zinc | = 0.0220 | " |
| ——— | ||
| ∴ Lead in 3 grams of the ore | = 0.1259 | " |
Equivalent to 4.20 per cent.
The same ore gave by separation of the lead with sulphuretted hydrogen, and conversion into sulphate, 4.16 per cent.
With fairly pure ores, free from sulphate, the assay may be made more quickly as follows: Dissolve 1 gram of the finely powdered ore by boiling gently with 40 c.c. of dilute hydrochloric acid for 15 minutes; cool; add a few drops of permanganate; neutralise with ammonia, add acetic acid and a little sodium acetate. Titrate with standard chromate.
This is based upon the brown coloration produced in very dilute solutions of lead by the action of a solution of sulphuretted hydrogen. The quantity of lead in the 50 c.c. of the assay solution must not much exceed 0.5 milligram, nor be less than 0.01. The sulphuretted hydrogen is used in the form of a solution, and is not bubbled through the assay. The principle of working is the same as previously described.
Standard Lead Solution.—Each c.c. of this should contain 0.1 milligram of lead. It is made by diluting 10 c.c. of the solution of lead nitrate, described under the volumetric process, to 1 litre.
Sulphuretted hydrogen water is made by passing a current of the washed gas into water till the latter is saturated.
Five c.c. of the sulphuretted hydrogen water are put into a Nessler tube, the measured portion of the assay solution added, and the whole diluted with water to the 50 c.c. mark. Into the standard Nessler tube the same amount of the sulphuretted hydrogen water is put, and diluted to nearly 50 c.c. The standard lead solution is then run in till the tints are equal. The assay solution must not contain much free acid, and if the conditions will allow it, may with advantage be rendered alkaline with ammonia. The chief cause of disturbance is the precipitation of lead sulphide forming a black turbid solution instead of a[Pg 219] brown clear one. This may be caused by using hot solutions or an excess of acid. Other metals precipitable by sulphuretted hydrogen must be absent as well as strong oxidising agents.
Effect of Varying Temperature.—The effect of increased temperature is to change the colour from brown to black, and to render the estimation difficult.
| 1 | c.c. | at 15° C. | showed the | colour of | 0.5 | c.c. | at 60° C. |
| 2 | " | " | " | " | 1.5 | " | at 60° C. |
| 3 | " | " | " | " | 5.0 | " | at 50° C. |
Effect of Varying Time.—The colour becomes lighter on standing: 2 c.c. on standing 10, 20, and 40 minutes became equal in colour to 1.7 c.c.
Effect of Acids and Ammonia.—Two c.c. of the solution with 2 c.c. of dilute hydrochloric acid became cloudy and equivalent to about 2.5 c.c.; and a similar result was got with 2 c.c. of dilute sulphuric acid. With 2 c.c. of dilute ammonia the solution became somewhat darker, or equal to 2.3 c.c.; but gave a very clear solution easy to compare.
Determination of Lead in Commercial Zinc.—Dissolve 0.1 gram of the metal in 1 c.c. of dilute nitric acid evaporates till a solid separates out, dilute to 100 c.c. with water, and take 20 c.c. for assay. A sample treated in this way required 2.4 c.c.; this multiplied by 5 gives 12.0 c.c., equivalent to 1.2 milligram of lead, or 1.2 per cent. By gravimetric assay the sample gave 1.10 per cent.
1. Thirty grams of galena gave on dry assay 21 grams of lead; and this, on cupellation, gave 15 milligrams of silver. Calculate the results in per cents. of lead and in ounces of silver to the ton of ore.
2. How many ounces of silver to the ton would be contained in the lead got from this ore if the loss in smelting is equal to that of the assay?
3. Having given you a sample of white lead freed from oil by washing with ether, how would you proceed to determine the percentage of lead in it?
Thallium is a rare metal, found in small quantities in some varieties of iron and copper pyrites, and in some lithia micas. It resembles lead in appearance. Its compounds resemble the salts of the alkalies in some respects; and, in others, those of the heavy metals.
It is detected by the green colour which its salts impart to the flame. This, when examined with the spectroscope, shows only one bright green line.[Pg 220]
It is separated and estimated by dissolving in aqua regia; converting into sulphate by evaporation with sulphuric acid; separating the second group of metals with sulphuretted hydrogen in the acid solution, boiling off the excess of the gas; nearly neutralising with carbonate of soda; and precipitating the thallium with an excess of potassic iodide. On allowing the liquid to stand for some time a bright yellow precipitate of thallous iodide separates out. This is collected on a weighed filter; washed with cold water, finishing off with alcohol; dried at 100° C., and weighed. The precipitate is thallous iodide TlI, and contains 61.6 per cent. of thallium.
Bismuth is nearly always found in nature in the metallic state; but occasionally it is met with as sulphide in bismuthine and as carbonate in bismutite. It is also found in some comparatively rare minerals, such as tetradymite, combined with tellurium, and associated with gold. In minute quantities it is widely distributed: it is a common constituent of most copper ores; hence it finds its way into refined copper, which is seldom free from it. It is occasionally met with in silver in sufficient quantity to interfere with the working qualities of that metal.
Bismuth compounds are used in medicine and in the manufacture of alloys. Bismuth possesses many useful properties. It has considerable commercial value, and sells at a high price.
The metal is brittle, breaks with a highly crystalline fracture, and has a characteristic reddish-yellow colour. It is almost insoluble in hydrochloric, but readily dissolves in nitric, acid; and gives, if the acid is in excess, a clear solution. Bismuth salts have a strong tendency to separate out as insoluble basic compounds; this is more especially true of the chloride which, on diluting with a large volume of water, becomes milky; the whole of the bismuth separating out. The nitrate, carbonate, and hydrate yield the oxide (Bi2O3) on ignition. This oxide closely resembles litharge. It combines with silica, forming fluid slags; and at a red heat is liquid enough to be absorbed by a cupel; in fact, bismuth may take the place of lead in cupellation. The metal itself is easily fusible, and may be separated from its ores by liquation.
The assay of bismuth by wet methods presents little difficulty, and is fairly accurate. The price of the metal is such that only methods which yield good results should be adopted; and, since bismuth is volatile at the temperature of the furnace, and is found mixed with ores not easy to flux, as also with metals which are not easily separated by the dry method, the dry assay can only be considered as having a qualitative value.[Pg 221]
By Liquation.—This is adapted to ores containing the bismuth as metal. Take 20 grams of the powdered ore and place in a crucible with a perforated bottom, put this crucible into another of about the same size and lute the joint. Lute on a cover, place in the furnace and heat to redness. The bismuth melts readily and drains into the lower crucible from which, when cold, it is taken and weighed.
By Fusion.—For fairly pure ores the process is as follows:—Take 20 grams of the ore and mix with 20 grams of fusion mixture, 10 grams of salt and 5 or 10 grams of potassium cyanide; place in a crucible, cover, and fuse at a moderate temperature for about fifteen minutes; pour; when cold detach the metal and weigh.
For coppery ores in which the metals are present as sulphides use the fluxes just given with 2 grams of charcoal (instead of the cyanide) and a little sulphur.
For coppery ores in which the metals are present as oxides, mix 20 grams of the ore with 10 grams of fusion mixture, 4 grams of salt, 4 grams of sulphur and 2 grams of charcoal; and fuse.
A considerable percentage of bismuth is lost in these assays; it is stated as being nearly 8 per cent. of the metal present.
Detection.—Bismuth is detected by dissolving the substance in nitric or hydrochloric acid and precipitating the diluted solution with sulphuretted hydrogen. The precipitated sulphides, after digesting with soda and washing, are dissolved in nitric acid and the solution boiled with ammonium carbonate. The precipitate is washed and then warmed with dilute sulphuric acid. The solution will contain the bismuth. Add a solution of potassium iodide in excess, and boil; a yellow or dark brown solution proves that bismuth is present. Another good test for small quantities of bismuth is to add tartaric acid to the solution to be tested, and then to make it alkaline with potash. Add a few c.c. of Schneider's liquid,[61] and heat. A brownish-black colour is produced by as little as one part of bismuth in 200,000 of solution. The test is not applicable in the presence of mercury, copper, or manganese.[Pg 222]
Compounds of bismuth fused with cyanide of potassium in a Berlin crucible readily give a globule of bismuth which is recognised by its appearance and fracture.
Solution and Separation.—The solution of bismuth compounds presents no difficulty. They are soluble in nitric acid or aqua regia, and, provided the solution is sufficiently acid, they remain dissolved. In separating it from other metals the solution is made up to about 100 c.c. and treated with a current of sulphuretted hydrogen. The bismuth comes down in a tolerably strong acid solution. The sulphide is decanted on to a filter and washed. It is next digested with ammonic sulphide; or, better (especially when other metals are present), dissolved in nitric acid, and treated with an excess of ammonia and a current of sulphuretted hydrogen. The precipitate is filtered off and evaporated to dryness with nitric acid. It is taken up with a few drops of sulphuric acid and a little water; and warmed and filtered, if necessary. The filtrate is nearly neutralised with ammonia; ammonium carbonate added in slight excess; and the liquid heated to boiling and filtered. The bismuth will be contained in the precipitate with perhaps traces of lead, antimony, tin, or sometimes iron from incomplete separation or washing. When only traces of a precipitate are got it must be tested. The bismuth precipitate is readily soluble in dilute nitric acid.
The bismuth having been separated and dissolved in nitric acid[62] is precipitated (after dilution) by the addition of carbonate of ammonium in slight excess, and boiling. The precipitate is filtered off, washed with hot water, dried, ignited, and weighed. The ignition should be performed carefully at not above a low red heat. The oxide which is formed has, at this temperature, a dark yellow or brown colour, and becomes yellow on cooling. It is bismuthic oxide (Bi2O3) and contains 89.65 per cent. of bismuth. Fusion with potassium cyanide at a temperature just sufficient to melt the salt reduces it to the metal which falls to the bottom and runs into a globule. The button of metal may be weighed, but it often sticks tenaciously to the bottom of the crucible. The precipitation with ammonic carbonate must not be made in a sulphate or chloride solution; since basic compounds would then be thrown down, and the result on weighing would either be too low (because of the volatilisation of the chloride), or too high (because of the retention of sulphuric acid).[Pg 223]
Bismuth compounds in a nitric acid solution are readily decomposed by the electric current, but the deposited bismuth is not coherent. It comes down in shaggy tufts which are difficult to wash and easy to oxidise.
There are two methods which have been proposed; one based on the precipitation as chromate and the estimation of the chromic acid; and the other on the precipitation as oxalate and subsequent titration with permanganate of potash. These offer little advantage over the easy gravimetric determination.
Bismuth iodide dissolves in excess of potassium iodide, forming a yellow-coloured solution, indistinguishable in colour from that given by iodine. The colour, however, is not removed by boiling or by sulphurous acid. Since none of the commoner metals give such a colour, and free iodine is easily separated by boiling, this method is specially suited for small determinations of bismuth.
It requires a solution of bismuth, made by dissolving 0.1 gram of bismuth in a drop or so of nitric acid, evaporating with a little sulphuric acid and diluting with water to 1 litre. 1 c.c. will contain 0.1 milligram of bismuth. And a solution of sulphurous acid, made by diluting 10 c.c. of the commercial acid to 1 litre with water.
The determination is made in the usual way: 50 c.c. of the prepared solution, which should not carry more than 0.75 milligram nor less than 0.01 milligram of bismuth, are placed in a Nessler tube and the colour compared with that observed in a similar tube containing water and potassium iodide on adding the standard solution of bismuth.
The assay solution is prepared by separating the bismuth with sulphuretted hydrogen, boiling the precipitate with nitric acid, and evaporating with sulphuric acid. Take up with water, add 10 or 20 c.c. of solution of potassium iodide, boil off any iodine liberated, dilute, filter, and make up to 100 c.c. According to the depth of colour take 10, 20, or 50 c.c. and transfer to the Nessler tube. Add a few c.c. of the solution of sulphurous acid. Into the other Nessler tube put as much potassium iodide solution as is contained in the assay tube, with sulphurous acid and water to within a few c.c. of the bulk. Then add the standard bismuth solution till the tints are equal.[Pg 224]
The student must be careful not to confuse the colour of the bismuth iodide with that of free iodine. If the yellow colour is removed by boiling and returns on standing it is due altogether to iodine; if it is lessened by the addition of a few drops of the dilute sulphurous acid, it is in part due to it. Hence the necessity of having a little free sulphurous acid in each tube. A strong solution must not be used, since it liberates iodine from potassium iodide.
The following experiments illustrate the effect of variation in the conditions of the assay:—
Effect of Varying Temperature.—At a higher temperature the colour is somewhat lessened.
| 1.0 | c.c. | at 15° C. | showed the colour of | 0.8 c.c. | at 70° C. |
| 2.5 | " | " | " | 2.0 | " |
| 5.0 | " | " | " | 5.0 | " |
Effect of Free Acid.—
| 2.5 | c.c. with | 5 c.c. of | nitric acid | equalled | 2.5 | c.c. |
| 5.0 | " | " | sulphuric acid | " | 5.0 | " |
Hydrochloric acid almost completely removes the colour, which, however, is restored by the addition of a few crystals of potassium iodide.
Effect of Alkalies.—Ammonia, soda, or potash destroys the colour, but it is restored on acidifying with nitric or sulphuric acid.
Effect of Ammonic Salts.—The following table shows the results after addition of ammonic salts:—
| C.c. present. | With 10 grams Ammonic Nitrate. | With 10 grams Ammonic Sulphate. | With 10 grams Ammonic Chloride. |
| 1.0 c.c. | 0.9 c.c. | 1.1 c.c. | — |
| 2.5 " | 2.5 " | 2.7 " | — |
| 5.0 " | 5.0 " | 5.5 " | — |
Ammonic chloride, like hydrochloric acid, removes the colour, which may be restored on the addition of more potassium iodide. Nitrates and sulphates do not thus interfere.
Effect of Foreign Salts.—Sodic hyposulphite almost completely removes the colour. Copper salts liberate iodine; but when this has been removed by boiling and the cuprous iodide has been filtered off there is no further interference. Dilute solutions of lead salts give no colour.[Pg 225]
1. A fusible alloy is made up of 8 parts of bismuth, 5 of lead, and 3 of tin. What weight of oxide of bismuth, Bi2O3, would you get on the analysis of 1 gram of it?
2. What weight of bismuth can be got from 2 grams of the subnitrate BiONO3.H2O?
3. How would you detect and separate arsenic, lead, and copper in a sample of bismuth?
Antimony occurs in the native state, but is rare; its common ore is antimonite, the sulphide (Sb2S3). Jamesonite and other sulphides of lead and antimony are frequently met with. Sulphide of antimony is also a constituent of fahlerz and of many silver ores.
Antimonite occurs generally in fibrous masses, has a lead-like metallic lustre, is easily cut with a knife, and melts in the flame of a candle.
Antimony itself has a very crystalline fracture, is brittle, and has a bluish-white colour. It is used in the preparation of alloys with lead and tin for the manufacture of type-metal. It is readily fusible, and imparts hardness and the property of taking a sharp cast to its alloys. It is practically insoluble in hydrochloric acid. On boiling with strong nitric acid it is converted into antimonic oxide (Sb2O5), which is a powder almost insoluble in this acid or in water, but which may be got into solution with difficulty by the prolonged action of hydrochloric and tartaric acids. Antimonic oxide is converted on ignition into the tetroxide (Sb2O4) with loss of oxygen. Antimony forms two series of salts, antimonious and antimonic; and advantage is taken of this in its determination volumetrically. Either sulphide of antimony yields antimonious chloride on boiling with hydrochloric acid, sulphuretted hydrogen being given off; and, in the case of antimonic sulphide, sulphur is deposited. Antimonious is converted into antimonic chloride by treatment with permanganate of potash in an acid solution. Antimonic chloride and potassium iodide react, forming antimonious chloride and free iodine. This latter may be got rid of by boiling. Sulphide of antimony is separated from the ore by liquation; this regulus is met with in commerce as "crude antimony."
An approximate determination of the amount of sulphide of antimony in an ore may be made by fusing and liquating in a luted double crucible in the manner described under bismuth.[Pg 226] This is unsatisfactory. The determination of metallic antimony in an ore is made either by fusion with potassium cyanide or by fusion with iron, as in the galena assay. Both methods yield poor results; and, where iron is used, it must be added in quantity only sufficient for desulphurising; this amounts to about 40 per cent. in pure ores. If the iron is in excess it alloys with the reduced antimony. If, on the other hand, it is insufficient, the metal will contain sulphur; or sulphide of antimony will be lost in the slag.
The following note, for which we are indebted to Mr. Bedford McNeill, A.R.S.M., gives a description of the method adopted in the commercial valuation of a parcel of antimony ore:—
The antimony smelter, when he wishes to determine the value of any parcel of ore—usually the sulphide—that may be offered for sale, practically has recourse to the smelting operation. That is, a quantity of 2 or 3 cwts. taken by his sampler having been obtained, he treats it under the immediate supervision of the foreman smelter as if it formed part of the ore in process of daily reduction at his works. He thus determines by actual trial the output which it may fairly be anticipated will be yielded by the bulk, and upon the result of this trial or assay, and the knowledge gained of the actual behaviour of the ore under treatment, he bases his tender, knowing that, should he secure the parcel, he may confidently expect a similar return.
Briefly, the process consists of the three ordinary operations of—
(a) Singling or removing most of the antimony from the ore;
(b) Doubling;
(c) Refining or "starring."
But in the assay sufficient information is generally given by the first two of these.
A new pot having been taken and made hot in the furnace, 40 or 45 lbs. of the ore is weighed in (the mineral from the necessities of sampling not exceeding walnut size); 1 to 3 lbs. of salt cake is now added to render the separation of the resulting sulphide of iron more easy, as also to assist in the fusion of the gangue; 20 to 25 lbs. of tin-plate scrap, beaten more or less into ball shape, is weighed, placed on the top of the ore and salt cake, and the whole brought to a state of fusion. The foreman from time to time takes notice of the behaviour of the ore under the working conditions. Ores that manifest a tendency to "boil" or "froth " require the admixture of other more sluggish mineral in order to render their reduction economically practicable.
After 1-1/4 to 1-1/2 hours (the time depending mainly on the temperature), the contents of the crucible are usually in a state of tranquil fusion. The pot is now lifted from the fire, and its[Pg 227] contents transferred to a conical iron mould, the empty pot being immediately put back into the fire, and the latter "mended" with sufficient coke for another run. The conical mould (when dealing with a "strange" ore, and the possibility of insufficient iron being present to satisfy the sulphur contents) is wiped inside with clay previous to pouring in the molten charge. Otherwise the mould itself will be attacked, and the contents after solidifying will require to be chiselled out piecemeal.
A further 40 lbs. of the ore is now charged into the crucible with iron as above; but before this second charge is ready to be drawn an inspection of the first may suggest the addition of either 3 or 5 lbs. more iron, or 5 or 10 lbs. more ore.
It is a good fault rather to aim at an excess of iron as tending to clean the ore from antimony, any of the latter that (from an insufficiency of iron) may be left in the slag from the first process being irretrievably lost; whereas, if the iron be in excess, that which is combined with the crude antimony resulting from the first process is easily got rid of by adding 3 to 5 lbs. or so of ore in the second process.
This latter, as practised for the determination of the value of a parcel of ore, consists in selecting two of the best quality singles, resulting from perhaps four or five trials as above, and running them down with a few pounds of salt cake, or a mixture of salt cake with American potash, and (as is generally necessary) a small addition of ore.
Upon the final result (confirmed perhaps on another pair of singles, and, judging from the total weight or output of the metal as calculated from the ore used in "singling," plus any added in the "doubling," the crystalline fracture and face of the metal, its colour, etc.) the price to be offered for the parcel of ore is fixed.
Detection.—The antimony, if any, being got into solution by treating the ore with hydrochloric acid or aqua regia may be detected by evaporating with hydrochloric acid, diluting, and filtering into the cover of a platinum crucible or (better) a platinum dish. A small lump of zinc is then added, and, if antimony is present, the dish will in a minute or so be stained black with a deposit of metallic antimony. This stain is removed by nitric, but not by hydrochloric, acid. The reaction is delicate and characteristic; arsenic under like conditions is evolved as arseniuretted hydrogen, and tin is deposited as metal on the zinc.
Solution.—Ores, &c., containing antimony are best opened up by boiling with hydrochloric acid or aqua regia; treatment with[Pg 228] nitric acid should be avoided wherever possible, since it forms antimonic acid, which is subsequently dissolved only with difficulty. Salts of antimony in solution have a tendency to form insoluble basic salts; so that care must be exercised in diluting. Compounds such as antimonite which are soluble in hydrochloric should be dissolved at once in that acid.
Separation.—To the solution add potash in excess and a little free sulphur, and pass a current of sulphuretted hydrogen for some minutes; allow to digest for an hour or so on a hot plate; filter; and wash the residue. Acidulate the filtrate with hydrochloric acid: the precipitate will contain the antimony (as Sb2S5), and possibly arsenic or tin. The precipitate is transferred to a beaker and boiled with hydrochloric acid; the solution is filtered off and diluted. Add a few crystals of tartaric acid, and pass a current of sulphuretted hydrogen for some time. The first flocculent precipitate will become denser, and render the filtering more easy. Transfer the precipitate (after washing free from chlorides) to a Berlin dish, and treat cautiously with fuming nitric acid. The action of this acid on the sulphide is very violent. Evaporate and ignite, transfer to a silver dish, and fuse with four or five times its weight of caustic soda, cool and extract with a little water, then add an equal volume of alcohol, and allow to stand overnight. Filter, wash with dilute alcohol. (The filtrate will contain the tin.) The residue contains the antimony as antimonate of soda, and is dissolved off the filter with hot dilute hydrochloric, with the help of a little tartaric, acid. The filtrate is now ready for the gravimetric determination.
Pass a current of sulphuretted hydrogen through the solution containing the antimony to which a little tartaric acid has been previously added. Pass the gas till the precipitate becomes dense, and the antimony is all down. The solution must not be too strongly acid. Filter off the precipitate, wash with hot water, dry in the water oven, transfer to a weighed porcelain dish, and cautiously treat with fuming nitric acid. Continue the action on the water bath till the sulphur and antimony are completely oxidised. Evaporate; ignite, gently at first, then strongly over the blast; cool, and weigh. The residue is a white infusible powder, and consists of antimony tetroxide, Sb2O4, containing 78.94 per cent. of the metal.
Determination of Antimony as Bigallate.—What appears to be a very good method has been worked out by M.A. Guyard, and is described in Crookes' Select Methods, p. 398.[Pg 229]
The antimony must be in solution as antimonious chloride, and must not be accompanied by an excess of hydrochloric acid. To ensure these conditions, the solution is treated with potassium iodide until no more iodine is evolved, and is then evaporated to remove the excess of hydrochloric acid. To the concentrated, and nearly neutral, solution a freshly-prepared solution of gallic acid is added in slight excess. A bulky white precipitate is formed that settles rapidly. The solution is diluted with hot water and washed by decantation. Then the precipitate is collected on a weighed double filter, washed once or twice with hot water, and dried at 100° C. The dried substance is antimony bigallate, and contains 40.85 per cent. of antimony. It should be completely soluble in ammonium sulphide. The solution in which the antimony is precipitated need not be quite free from other metals.
This is based on the reduction of antimonic chloride (SbCl5) to antimonious (SbCl3) by the action of potassium iodide in strong hydrochloric acid solution.[63] Iodine is at the same time liberated, and the amount of antimony reduced is got at by titrating with sodium hyposulphite, which measures the iodine set free.
The standard solution of sodium hyposulphite is made by dissolving 41.32 grams of the salt (Na2S2O3.5H2O) in water, and diluting to 1 litre. One hundred c.c. will be equivalent to about 1 gram of antimony.
It is standardised with the help of a solution of antimony made as follows:—Weigh up 5 grams of powdered antimony, transfer to a flask, and cover with 50 c.c. of hydrochloric acid; boil, and add nitric acid (5 or 10 drops at a time) until the metal is dissolved. Allow the action of the nitric acid to cease before adding more. Boil down to a small bulk, add 250 c.c. of hydrochloric acid, and dilute to nearly 1 litre. Warm until any precipitate which has formed is redissolved; allow to cool slowly, and run in from a pipette a weak solution of permanganate until a faint brown colour is produced. Dilute to exactly 1 litre; 100 c.c. contain 0.5 gram of antimony as antimonic chloride.
In standardising, take 50 c.c. of the antimony solution, and transfer to a flask; add 2 grams of potassium iodide crystals, and when dissolved, after standing a few minutes, run in the solution of "hypo" from an ordinary burette until the greater part of the iodine has been reduced. Add a few drops of starch solution, and continue the addition of the "hypo" until the muddy-green colour[Pg 230] changes to a clear brownish-yellow. The solution must be shaken after each addition of the "hypo."
In determining antimony in ore, weigh up 0.5 to 1 gram, and dissolve in hydrochloric acid with, if necessary, the help of chlorate of potash. The antimony is separated as sulphide, redissolved in hydrochloric acid, and oxidised with a crystal of chlorate of potash. Chlorine is boiled off, and the solution diluted with an equal bulk of water. To the clear cold solution potassium iodide is added, and after a few minutes the liberated iodine is titrated with "hypo," as already described. The method only yields satisfactory results when the standard and assay are carried out alike.
[50] "Modern American Methods of Copper Smelting" (Dr. Peters).
[51] "Journal of the Society of Chemical Industry," vol. v. No. 2.
[52] Lead when present is precipitated on the spiral in the form of a dark powder of dioxide (PbO2). Manganese is also thrown down on the spiral as dioxide (MnO2), the solution at the same time becomes violet from the formation of permanganic acid.
[53] See the method given under Examination of Commercial Copper.
[54] CuSO4 + 4KCy = 2KCy.CuCy2 + K2SO4.
[55] 2CuSO4 + 3KCy + Am2O = Cu2Cy2 + Am2SO4 + K2SO4 + KCyO.
[56] 2CuSO4 + 4KI = Cn2I2 + 2I + 2K2SO4.
[57] 2Na2S2O3 + 2I = 2NaI + Na2S4O6.
[58] For further information, see Appendix B., and a paper by J.W. Westmoreland, Journal of the Society of Chemical Industry, vol. v. p. 48.
[59]
3Cu2O + 6AgNO3 + 3H2O
= 2Cu2H3O3NO3 + 2Cu(NO3)2 + 6Ag.
(Insoluble basic salt.)
[60] K2CrO4 + Pb(NO3)2 = PbCrO4 + 2KNO3
[61] Made by dissolving 12 grams of tartaric acid and 4 grams of stannous chloride in water, and adding potash solution till it is alkaline. The solution should remain clear on heating to 60° or 70° C.
[62] It must be remembered that arsenate of bismuth is completely insoluble in this acid.
[63] SbCl5 + 2KI = I2 + SbCl3 + 2KCl.
Iron rusts or oxidises very readily, and, consequently, is rarely found in the metallic state in nature; such native iron as is found being generally of meteoric origin or imbedded in basalt and other igneous rocks. It chiefly occurs as oxide, as in magnetite, hæmatite, and in the brown iron ores and ochres. Chalybite, which is carbonate of iron, is an ore of great importance. Iron is found combined with sulphur in pyrrhotine and pyrites, and together with arsenic in mispickel. It is a common constituent of most rocks, imparting to them a green, black, or brown colour; and is present, either as an essential part or as an impurity, in most substances.
The chemistry of iron is somewhat complicated by the existence of two oxides, each of which gives rise to a well-marked series of compounds. Those derived from the lower oxide, known as ferrous salts, are generally pale and greenish. Ferric salts are derived from the higher oxide, and are generally red, brown, or yellow. The existence of these two well-marked families of salts renders the assay of iron comparatively easy, for the quantity of iron present in a solution can be readily measured by the amount of oxidising or reducing agent required to convert it from the one state into the other—that is, from ferrous to ferric, or from ferric to ferrous, as the case may be.
In the red and brown iron ores and ochres ferric iron is present; in chalybite the iron is in the ferrous state; and in magnetite it is present in both forms. Traces of iron in the ferrous state may be found (even in the presence of much ferric iron) by either of the following tests:—
1. Ferricyanide of potassium gives a blue precipitate or green coloration; with ferric salts a brown colour only is produced.
2. A solution of permanganate of potassium is decolorised by a ferrous salt, but not by a ferric one.
Traces of ferric iron can be detected (even in the presence of much ferrous iron) by the following tests:—
(1) By the brown or yellow colour of the solution, especially when hot.
(2) By giving a pink or red coloration with sulphocyanide of potassium.
Substances containing oxide of iron yield the whole of the iron as metal when fused at a high temperature with charcoal and suitable fluxes. The metal, however, will contain varying proportions of carbon and other impurities, and its weight can only afford a rough knowledge of the proportion of the metal in the ore. There are two or three methods of dry assay for iron, but they are not only inexact, but more troublesome than the wet methods, and need not be further considered. Chalybite and the hydrated oxides dissolve very readily in hydrochloric acid; hæmatite and magnetite dissolve with rather more difficulty. Iron itself, when soft, is easily soluble in dilute hydrochloric, or sulphuric, acid. Pyrites, mispickel, &c., are insoluble in hydrochloric acid, but they are readily attacked by nitric acid. Certain minerals, such as chrome iron ore, titaniferous iron ore, and some silicates containing iron, remain in the residue insoluble in acids. Some of these yield their iron when attacked with strong sulphuric acid, or when fused with the acid sulphate of potash. Generally, however, it is better in such stubborn cases to fuse with carbonate of soda, and then attack the "melt" with hydrochloric acid.
When nitric acid, or the fusion method, has been used, the metal will be in solution in the ferric state, no matter in what condition it existed in the ore. But with dilute hydrochloric or sulphuric acid it will retain its former degree of oxidation. Hydrochloric acid, for example, with chalybite (ferrous carbonate) will give a solution of ferrous chloride; with hæmatite (ferric oxide) it will yield ferric chloride; and with magnetite (ferrous and ferric oxides) a mixture of ferrous and ferric chlorides. Metallic iron yields solutions of ferrous salts. It is convenient to speak of the iron in a ferrous salt as ferrous iron, and when in the ferric state as ferric iron. Frequently it is required to determine how much of the iron exists in an ore in each condition. In such cases it is necessary to keep off the air whilst dissolving; the operation should, therefore, be performed in an atmosphere of carbonic acid.
Separation.—The separation of the iron from the other substances is as follows:—Silica is removed by evaporating the acid solution, and taking up with acid, as described under Silica; the whole of the iron will be in solution. The metals of Groups I. and II. are removed by passing sulphuretted hydrogen,[Pg 233] and at the same time the iron will be reduced to the ferrous state. The solution should be filtered into a 16 oz. flask, boiled to get rid of the gas, and treated (whilst boiling) with a few drops of nitric acid, in order to convert the whole of the iron into the ferric state. When this condition is arrived at, an additional drop of nitric acid causes no dark coloration. The boiling must be continued to remove nitrous fumes. Next add caustic soda solution until the colour of the solution changes from yellow to red. The solution must be free from a precipitate; if the soda be incautiously added a permanent precipitate will be formed, in which case it must be redissolved with hydrochloric acid, and soda again, but more cautiously, added. After cooling, a solution of sodium acetate is added until the colour of the solution is no longer darkened. The solution, diluted to two-thirds of the flaskful with water, is heated to boiling. Long-continued boiling must be avoided. The precipitate is filtered quickly through a large filter, and washed with hot water containing a little acetate of soda.
The precipitate will contain all the iron and may also contain alumina, chromium, titanium, as well as phosphoric, and, perhaps, arsenic acids.[64]
Dissolve the precipitate off the filter with dilute sulphuric acid, avoiding excess, add tartaric acid and then ammonia in excess. Pass sulphuretted hydrogen, warm, and allow the precipitate to settle. Filter and wash with water containing a little ammonic sulphide.
Dissolve the precipitate in dilute hydrochloric acid; peroxidise with a few drops of nitric acid and boil, dilute to about 200 c.c., add ammonia (with constant stirring) till the liquid smells of it, and heat to boiling. Wash as much as possible by decantation with hot water. Transfer to the filter, and wash till the filtrate gives no indication of soluble salts coming through. The filtrate must be colourless and clear. The wet precipitate is very bulky, of a dark-brown colour and readily soluble in dilute acids, but insoluble in ammonia and dilute alkalies. When thrown down from a solution containing other metals it is very apt to carry portions of these with it, even when they are by themselves very soluble in ammoniacal solutions. It must be dried and ignited, the filter paper being burnt separately and its ash added. When further ignition ceases to cause a loss of weight, the residue is ferric oxide (Fe2O3), which contains 70 per cent. of iron. The weight of iron therefore can be calculated by multiplying the weight of oxide obtained by 0.7.[Pg 234]
The presence of ammonic chloride causes loss of iron during the ignition, and organic matter causes an apparent loss by reducing the iron to a lower state of oxidation. When the iron in the solution much exceeds 0.2 gram the volumetric determination is generally adopted, as the bulkiness of the precipitate of ferric hydrate makes the gravimetric method very inconvenient.
As already explained these are based on the measurement of the volume of a reagent required to bring the whole of the iron from the ferrous to the ferric state (oxidation), or from the ferric to the ferrous (reduction). Ferrous compounds are converted into ferric by the action of an oxidising agent in the presence of an acid. Either permanganate or bichromate of potash is generally used for this purpose.[65]
Ferric compounds are reduced to ferrous by the action of:—
(1) Stannous chloride;
(2) Sulphuretted hydrogen;
(3) Sodium sulphite; or
(4) Zinc.[66]
The processes, then, may be divided into two kinds, one based on oxidation and the other on reduction. In each case the titration must be preceded by an exact preparation of the solution to be assayed in order that the iron may be in the right state of oxidation.
These consist of three operations:—
(1) Solution of the ore;
(2) Reduction of the iron to the ferrous state; and
(3) Titration.
Solution.—The only point to be noticed concerning the first operation (in addition to those already mentioned) is that nitric acid must be absent. If nitric acid has been used, evaporate to dryness, of course without previous dilution; add hydrochloric or sulphuric acid, and boil for five or ten minutes. Dilute with water to about 100 c.c., and warm until solution is complete.
The reduction is performed by either of the following methods:[Pg 235]—
1. With Stannous Chloride.—Fill a burette with a solution of stannous chloride,[67] and cautiously run the liquid into the hot assay solution (in which the iron is present as chloride) until the colour is discharged. A large excess of the stannous chloride must be avoided. Then add 5 c.c. of a 2-1/2 per cent. solution of mercuric chloride, this will cause a white precipitate (or a grey one if too large an excess of the stannous chloride has been added). Boil till the solution clears, cool, dilute, and titrate.
2. With Sulphuretted Hydrogen.—Cool the solution and pass through it a current of washed sulphuretted hydrogen till the liquid smells strongly of the gas after withdrawal and shaking. A white precipitate of sulphur will be formed, this will not interfere with the subsequent titration provided it is precipitated in the cold. If, however, the precipitate is coloured (showing the presence of the second group metals), or if the precipitation has been carried out in a hot solution, it should be filtered off. Boil the solution until the sulphuretted hydrogen is driven off; this may be tested by holding a strip of filter paper dipped in lead acetate solution in the steam issuing from the flask. The presence of sulphuretted hydrogen should be looked for rather than its absence. It is well to continue the boiling for a few minutes after the gas has been driven off. Cool and titrate.
3. With Sodium Sulphite.—Add ammonia (a few drops at a time) until the precipitate first formed redissolves with difficulty. If a permanent precipitate is formed, redissolve with a few drops of acid. To the warm solution add from 2 to 3 grams of sodium sulphite crystals. The solution will become strongly coloured, but the colour will fade away on standing for a few minutes in a warm place. When the colour is quite removed, add 20 c.c. of dilute sulphuric acid, and boil until the steam is quite free from the odour of sulphurous acid. Cool and titrate.
4. With Zinc.—Add about 10 grams of granulated zinc; if the hydrogen comes off violently add water; if, on the other hand, the action is very slow, add sufficient dilute sulphuric acid to keep up a brisk effervescence. The reduction is hastened by warming, and is complete when the solution is quite colourless and a drop of the liquid tested with sulphocyanate of potassium gives no reaction for ferric iron. Filter through "glass wool" or quick filtering paper. The zinc should be still giving off gas rapidly, indicating a freely acid solution; if not, acid must be added. Wash with water rendered acid. Cool and titrate.
With regard to the relative advantages of the different methods they may be roughly summed up as follows:—The stannous[Pg 236] chloride method has the advantage of immediately reducing the ferric iron whether in hot or cold solution and under varied conditions in regard to acidity, but has the disadvantage of similarly reducing salts of copper and antimony, which, in a subsequent titration, count as iron. Moreover, there is no convenient method of eliminating any large excess of the reagent that may have been used; and, consequently, it either leaves too much to the judgment of the operator, or entails as much care as a titration. Students generally get good results by this method.
The sulphuretted hydrogen method also has the advantage of quick reduction under varying conditions, and the further one of adding nothing objectionable to the solution; in fact it removes certain impurities. The disadvantages are the necessity for boiling off the excess of the gas, and of filtering off the precipitated sulphur, although this last is not necessary if precipitated cold. The tendency with students is to get high results. The sodium sulphite method has the advantages of being clean and neat, and of requiring no nitration. On the other hand it requires practice in obtaining the best conditions for complete reduction; and, as with sulphuretted hydrogen, there is the necessity for boiling off the gas, while there is no simple and delicate test for the residual sulphurous acid. In addition, if an excess of sodium sulphite has been used and enough acid not subsequently added, the excess will count as iron. Students generally get low results by this method.
The advantages of the zinc method are, that it is easily worked and that the excess of zinc is readily removed by simply filtering. The disadvantages are the slowness[68] with which the last portions of ferric iron are reduced, the danger of loss by effervescence, the precipitation of basic salts, and, perhaps, of iron, and the loading of the solution with salts of zinc, which in the titration with bichromate have a prejudicial effect. The tendency in the hands of students is to get variable results, sometimes low and sometimes high.
Generally speaking, the sulphuretted hydrogen and sodium sulphite methods are to be preferred. Carefully worked each method will yield good results.
The titration may be done with a standard solution of (1) permanganate of potash, or (2) bichromate of potash.
1. With Permanganate of Potash.—Prepare a standard solution by dissolving 2.82 grams of the salt and diluting to one litre. The strength of this should be 100 c.c. = 0.5 gram of iron, but it varies slightly, and should be determined (and afterwards[Pg 237] checked every two or three weeks) by weighing up 0.2 gram of iron wire, dissolving in 10 c.c. of dilute sulphuric acid, diluting to about 100 c.c., and titrating.
The standard solution must be put in a burette with a glass stopcock, as it attacks india-rubber. The assay should be contained in a pint flask, and be cooled before titrating. The standard solution must be run in until a pinkish tinge permeates the whole solution; this must be taken as the finishing point. When certain interfering bodies are present this colour quickly fades, but the fading must be ignored. With pure solutions the colour is fairly permanent, and a single drop of the potassium permanganate solution is sufficient to determine the finishing point.
2. With Bichromate of Potash.—Prepare a standard solution by dissolving 4.39 grams of the powdered and dried salt in water, and diluting to 1 litre. This solution is permanent, its strength is determined by dissolving 0.2 gram of iron wire in 10 c.c. of dilute sulphuric acid, diluting to about a quarter of a litre, and titrating.
Also prepare a test solution by dissolving 0.1 gram of ferricyanide of potassium in 100 c.c. of water. This solution does not keep well and must be freshly prepared.
An ordinary burette is used. The assay is best contained in a glazed earthenware dish, and may be titrated hot or cold. To determine the finishing point, place a series of drops of the ferricyanide solution on a dry white glazed plate. The drops should be of about the same size and be placed in lines at fairly equal distances. The bichromate is run in, in a steady stream, the assay solution being continuously stirred until the reaction is sensibly slackened. Then bring a drop of the assay with the stirrer in contact with one of the test drops on the plate. The standard can be safely run in 1 c.c. at a time, so long as the test drop shows signs of a precipitate. When only a coloration is produced run in cautiously a few drops at a time so long as two drops of the assay gives with the test a colour which is even faintly greener than two drops of the assay solution placed alongside. The finishing point is decided and practically permanent, although it demands a little practice to recognise it. The titration with permanganate of potassium has the advantage of a more distinct finishing point and easier mode of working; its application, however, is somewhat limited by the disturbing effects of hydrochloric acid. The bichromate method has the advantage of a standard solution which does not alter in strength, and the further one of being but little affected by altering conditions of assay. Hydrochloric acid has practically no effect on it. Both methods give accurate results and are good examples of volumetric methods.[Pg 238]
The following results illustrate the extent to which the methods may be relied on; and the influence which the various conditions of experiment have on the assay.
Solutions of ferrous sulphate and of ferrous chloride were made containing 0.5 gram of iron in each 100 c.c., thus corresponding to the standard solutions of permanganate and bichromate of potassium. These last were prepared in the way already described. The solution of ferrous sulphate was made by dissolving 5.01 grams of iron wire in 100 c.c. of dilute sulphuric acid and diluting to 1 litre. A similar solution may be made by dissolving 24.82 grams of pure ferrous sulphate crystals in water, adding 100 c.c. of dilute sulphuric acid, and diluting to 1 litre.
Rate of Oxidation by Exposure to Air.—This is an important consideration, and if the rate were at all rapid would have a serious influence on the manner of working, since exclusion of air in the various operations would be troublesome. 20 c.c. of the solution of ferrous sulphate were taken in each experiment, acidified with 10 c.c. of dilute sulphuric acid, and diluted to 100 c.c. The solution was exposed, cold, in an open beaker for varying lengths of time, and titrated with permanganate of potassium.
| Time exposed | 1 hour | 1 day | 2 days | 3 days |
| c.c. required | 19.2 | 19.1 | 19.0 | 19.0 |
These results show that the atmospheric oxidation in cold solutions is unimportant. With boiling solutions the results are somewhat different; a solution which at the outset required 20 c.c. of permanganate of potassium, after boiling for an hour in an open beaker (without any precautions to prevent oxidation), water being added from time to time to replace that lost by evaporation, required 19.2 c.c. If the solution be evaporated to dryness the oxidising power of concentrated sulphuric acid comes into play, so that very little ferrous iron will be left. A solution evaporated in this way required only 2.2 c.c. of permanganate of potassium.
Effect of Varying Temperature.—In these experiments the bulk was in each case 100 c.c., and 10 c.c. of dilute sulphuric acid were present. The permanganate required by
| 1 | c.c. | of ferrous sulphate was, | at 15° | 1.0 c.c., | and at 70° | 1.1 | c.c. |
| 10 | " | " | " | 9.7 | " | 9.8 | " |
| 100 | " | " | " | 97.7 | " | 96.8 | " |
The lower result with the 100 c.c. may be due to oxidation from exposure.[Pg 239]
Effect of Varying Bulk.—The following experiments show that considerable variations in bulk have no practical effect. In each case 20 c.c. of ferrous sulphate solution and 10 c.c. of dilute acid were used.
| Bulk of assay | 30 | c.c. | 100 | c.c. | 500 | c.c. | 1000 | c.c. |
| Permanganate required | 20.0 | " | 20.0 | " | 20.2 | " | 20.5 | " |
Effect of Free Sulphuric Acid.—Free acid is necessary for these assays; if there is an insufficiency, the assay solution, instead of immediately decolorising the permanganate, assumes a brown colour. The addition of 10 c.c. of dilute sulphuric acid suffices to meet requirements and keep the assay clear throughout. The following experiments show that a considerable excess of acid may be used without in the least affecting the results. In each case 20 c.c. of ferrous sulphate were used.
| Dilute sulphuric acid | 1.0 | c.c. | 5.0 | c.c. | 10.0 | c.c. | 20.0 | c.c. | 50.0 | c.c. | 100.0 | c.c. |
| Permanganate required | 19.3 | " | 19.3 | " | 19.3 | " | 19.3 | " | 19.3 | " | 19.3 | " |
Effect of Foreign Salts.—When the assay has been reduced with zinc varying quantities of salts of this metal pass into solution, the amount depending on the quantity of acid and iron present. Salts of sodium or ammonium may similarly be introduced. It is essential to know by experiment that these salts do not exert any effect on the titration. The following series of experiments show that as much as 50 grams of zinc sulphate may be present without interfering.
| Zinc sulphate present | 0 gram | 1 gram | 10 grams | 50 grams |
| Permanganate required | 19.3 c.c. | 19.3 c.c. | 19.3 c.c. | 19.3 c.c. |
Magnesium, sodium, and ammonium salts, are equally without effect.
| Ammonic sulphate present | 0 gram | 1 gram | 10 grams |
| Permanganate required | 19.3 c.c. | 19.2 c.c. | 19.3 c.c. |
| Sodic sulphate present | 0 gram | 1 gram | 10 grams |
| Permanganate required | 19.3 c.c. | 19.3 c.c. | 19.3 c.c. |
| Magnesic sulphate present | 0 gram | 1 gram | 10 grams |
| Permanganate required | 19.3 c.c. | 19.3 c.c. | 19.3 c.c. |
Effect of Varying Amounts of Iron.—It is important to know within what limits the quantity of iron in an assay may safely vary from that used in standardising. In the following experiments the conditions as to bulk, acidity, and mode of working were the same as before:—
| Ferrous sulphate solution taken | 1 | c.c. | 10 | c.c. | 20 | c.c. | 50 | c.c. | 100 | c.c. |
| Permanganate required | 1.0 | " | 9.7 | " | 19.6 | " | 48.9 | " | 97.7 | " |
The ferrous sulphate solution is here a little weaker than that of the permanganate of potassium, but the results show that the permanganate required is proportional to the iron present.
Titrations in Hydrochloric Solutions.—These are less satisfactory than those in sulphuric solutions, since an excess of hydrochloric acid decomposes permanganate of potassium, evolving chlorine, and since the finishing point is indicated, not by the persistence of the pink colour of the permanganate, but by a brown coloration probably due to perchloride of manganese. Nevertheless, if the solution contains only from 5 to 10 per cent. of free hydrochloric acid (sp. g. 1.16) the results are the same as those obtained in a sulphuric acid solution. Equal weights (0.1 gram) of the same iron wire required exactly the same quantity of the permanganate of potassium solution (20 c.c.) whether the iron was dissolved in dilute sulphuric or dilute hydrochloric acid. The following series of experiments are on the same plan as those given above with sulphuric acid solutions. A solution of ferrous chloride was made by dissolving 5.01 grams of iron wire in 50 c.c. of dilute hydrochloric acid and diluting to 1 litre. The dilute hydrochloric acid was made by mixing equal volumes of the acid (sp. g. 1.16) and water.
Rate of Atmospheric Oxidation.—20 c.c. of the ferrous chloride solution were acidified with 10 c.c. of the dilute hydrochloric acid and diluted to 100 c.c. This solution was exposed cold in open beakers.
| Time exposed | — | 1 hour | 1 day | 2 days | 3 days |
| Permanganate required | 19.6 c.c. | 19.6 c.c. | 19.5 c.c. | 19.4 c.c. | 19.5 c.c. |
Similar solutions boiled required, before boiling, 20 c.c.; after boiling for one hour, replacing the water as it evaporated, 19.3 c.c.; and after evaporation to a paste and redissolving, 17.0 c.c.
Effect of Varying Temperature.—Solutions similar to the last were titrated and gave the following results:—
| Temperature | 15° | 30° | 50° | 70° |
| Permanganate required | 19.8 c.c. | 19.6 c.c. | 19.5 c.c. | 19.4 c.c. |
Effect of Varying Bulk.—As before, 20 c.c. of the iron solution, and 10 c.c. of the dilute acid were diluted to the required volumes and titrated.
| Bulk | 30 c.c. | 100 c.c. | 500 c.c. | 1000 c.c. |
| Permanganate required | 20.4 " | 20.3 " | 20.8 " | 21.5 " |
The variation due to difference in bulk here, although only equal to an excess of 0.7 milligram of iron for each 100 c.c. of dilution, are about three times as great as those observed in a sulphuric acid solution.[Pg 241]
Effect of Free Hydrochloric Acid.—In these experiments 20 c.c. of the ferrous chloride solution were used with varying quantities of acid, the bulk of the assay in each case being 100 c.c.
| Dilute acid present | 5 c.c. | 10 c.c. | 50 c.c. | 100 c.c. |
| Permanganate required | 20.2 " | 20.2 " | 20.5 " | 21.0 " |
The last had a very indistinct finishing point, the brown coloration being very evanescent. The effect of the acid is modified by the presence of alkaline and other sulphates, but not by sulphuric acid. Repeating the last experiment we got—
| Without further addition | 21.0 | c.c. | |||
| With | 100 | c.c. | of dilute sulphuric acid | 22.0 | " |
| " | 10 | grams | ammonic sulphate | 20.5 | " |
| " | 10 | " | sodic sulphate | 20.0 | " |
| " | 10 | " | magnesium sulphate | 20.4 | " |
| " | 10 | " | manganese sulphate | 20.2 | " |
The results with these salts, in counteracting the interference of the acid, however, were not a complete success, since the end-reactions were all indistinct, with the exception, perhaps, of that with the manganese sulphate.
Effect of Varying Amounts of Iron.—In these experiments the bulk of the assay was 100 c.c., and 10 c.c. of acid were present.
| Ferrous chloride used | 1 c.c. | 10 c.c. | 20 c.c. | 50 c.c. | 100 c.c. |
| Permanganate required | 1.1 " | 10.3 " | 20.3 " | 50.4 " | 100.1 " |
In making himself familiar with the permanganate of potassium titration, the student should practise by working out a series of experiments similar to the above, varying his conditions one at a time so as to be certain of the cause of any variation in his results. He may then proceed to experiment on the various methods of reduction.
A solution of ferric chloride is made by dissolving 5.01 grams of iron wire in 50 c.c. of hydrochloric acid (sp. g. 1.16), and running from a burette nitric acid diluted with an equal volume of water into the boiling iron solution, until the liquid changes from a black to a reddish-yellow. About 4.5 c.c. of the nitric acid will be required, and the finishing point is marked by a brisk effervescence. The solution of iron should be contained in an evaporating dish, and boiled briskly, with constant stirring. There should be no excess of nitric acid. Boil down to about half its bulk; then cool, and dilute to one litre with water. Twenty c.c. of this solution diluted to 100 c.c. with water, and acidified with 10 c.c. of dilute hydrochloric acid, should not decolorise any of the permanganate of potassium solution; this shows the absence of ferrous salts. And 20 c.c. of the same solution, boiled with[Pg 242] 20 c.c. of the ferrous sulphate solution, should not decrease the quantity of "permanganate" required for the titration of the ferrous sulphate added. In a series of experiments on the various methods of reduction, the following results were got. The modes of working were those already described.
(1) With Stannous Chloride.—Twenty c.c. of the ferric chloride solution required, after reduction with stannous chloride, 20 c.c. of "permanganate." Fifty c.c. of a solution of ferrous chloride, which required on titration 49.8 c.c. of "permanganate," required for re-titration (after subsequent reduction with stannous chloride) 50 c.c. of the permanganate solution.
(2) With Sulphuretted Hydrogen.—Two experiments with this gas, using in each 20 c.c. of the ferric chloride solution, and 10 c.c. of hydrochloric acid, required (after reduction) 20.2 c.c. and 20.1 c.c. of "permanganate." Repeating the experiments by passing the gas through a nearly boiling solution, but in other respects working in the same way, 21.3 c.c. and 21.6 c.c. of the permanganate solution were required. The sulphur was not filtered off in any of these. In another experiment, in which 50 c.c. of the ferrous sulphate solution were titrated with "permanganate," 48 c.c. of the latter were required. The titrated solution was next reduced with sulphuretted hydrogen, brought to the same bulk as before, and again titrated; 47.9 c.c. of the permanganate of potassium solution were required.
(3) With Sodium Sulphite.—Twenty c.c. of the ferric chloride solution, reduced with sodium sulphite, required 19.9 c.c. of "permanganate." In one experiment 50 c.c. of the ferrous sulphate solution were titrated with "permanganate"; 49.3 c.c. of the last-mentioned solution were required. The titrated solution was reduced with sodium sulphite, and again titrated; it required 49.2 c.c. of the permanganate of potassium solution.
(4) With Zinc.—Twenty c.c. of the ferric chloride solution, reduced with zinc and titrated, required 20.8 c.c. of "permanganate." Fifty c.c. of a solution of ferrous sulphate which required 49.7 c.c. of "permanganate," required for re-titration, after reduction with zinc, 49.7 c.c.
The student should next practise the titration with bichromate, which is more especially valuable in the estimation of hydrochloric acid solutions. The following experiments are on the same plan as those already given. In each experiment (except when otherwise stated) there were present 20 c.c. of the ferrous chloride solution, and 10 c.c. of dilute hydrochloric acid, and the bulk was 300 c.c.
Effect of Varying Temperature.—The quantities of the bichromate of potassium solution required were as follows:[Pg 243]—
| Temperature | 15° | 30° | 70° | 100° |
| Bichromate required | 20.2 c.c. | 20.3 c.c. | 20.3 c.c. | 20.4 c.c. |
Effect of Varying Bulk.—
| Bulk | 50 c.c. | 100 c.c. | 200 c.c. | 500 c.c. | 1000 c.c. |
| Bichromate required | 20.4 " | 20.4 " | 20.4 " | 20.5 " | 20.8 " |
Effect of Varying Acid.—In these, variable quantities of dilute hydrochloric acid were used.
| Acid present | 10 c.c. | 50 c.c. | 100 c.c. |
| Bichromate required | 20.3 " | 20.3 " | 20.2 " |
Effect of Foreign Salts.—The effect of the addition of 10 grams of crystallized zinc sulphate was to decrease the quantity of "bichromate" required from 20.3 c.c. to 20.1 c.c., but the colour produced with the test-drop was very slight at 18.5 c.c., and with incautious work the finishing point might have been taken anywhere between these extremes. Zinc should not be used as a reducing agent preliminary to a "bichromate" titration. Ten grams of ammonic sulphate had the effect of rendering the finishing point faint for about 0.5 c.c. before the titration was finished, but there was no doubt about the finishing point when allowed to stand for a minute. The student should note that a titration is not completed if a colour is developed on standing for five or ten minutes. Ten grams of sodic sulphate had no effect; 20.3 c.c. were required.
Effect of Varying Iron.—The results are proportional, as will be seen from the following details:—
| Ferrous chloride present | 1.0 c.c. | 10.0 c.c. | 20.0 c.c. | 50.0 c.c. | 100.0 c.c. |
| Bichromate required | 1.0 " | 10.2 " | 20.3 " | 51.0 " | 102.3 " |
The student may now apply these titrations to actual assays of minerals. The following examples will illustrate the mode of working and of calculating the results:—
Determination of Iron in Chalybite.—Weigh up 1 gram of the dry powdered ore, and dissolve in 10 c.c. of dilute sulphuric acid and an equal volume of water with the aid of heat. Avoid evaporating to dryness. Dilute and titrate. The result will give the percentage of iron existing in the ore in the ferrous state. Some ferric iron may be present. If it is wished to determine this also, add (in dissolving another portion) 10 c.c. of dilute hydrochloric acid to the sulphuric acid already ordered, and reduce the resulting solution before titrating. By dissolving and titrating (without previous reduction) one has a measure of the ferrous iron present; by dissolving, reducing, and then titrating, one can[Pg 244] measure the total iron; and as the iron exists in only two conditions, the total iron, less the ferrous iron, is the measure of the ferric iron.
Determination of Iron in Brown or Red Ores or Magnetite.—Weigh up 0.5 gram of the ore (powdered and dried at 100° C.), and dissolve in from 10 to 20 c.c. of strong hydrochloric acid, boiling until all is dissolved, or until no coloured particles are left. Dilute, reduce, and titrate.
Determination of Iron in Pyrites.—Weigh up 1 gram of the dry powdered ore, and place in a beaker. Cover with 10 c.c. of strong sulphuric acid, mix well by shaking, and place on the hot plate without further handling for an hour or so until the action has ceased. Allow to cool, and dilute to 100 c.c. Warm until solution is complete. Reduce and titrate.
Determination of Iron in Substances Insoluble in Acids.—Weigh up 1 gram of the ore, mix with 5 or 6 grams of carbonate of soda and 0.5 gram of nitre by rubbing in a small mortar, and transfer to a platinum crucible. Clean out the mortar by rubbing up another gram or so of soda, and add this to the contents of the crucible as a cover. Fuse till tranquil. Cool. Extract with water. If the ore carries much silica, evaporate to dryness with hydrochloric acid to separate it. Re-dissolve in hydrochloric acid, and separate the iron by precipitating with ammonia and filtering. If only a small quantity of silica is present, the aqueous extract of the "melt" must be filtered, and the insoluble residue washed and dissolved in dilute hydrochloric acid. Reduce and titrate.
A convenient method of at once separating iron from a solution and reducing it, is to add ammonia, pass sulphuretted hydrogen through it, filter, and dissolve the precipitate in dilute sulphuric acid. The solution, when boiled free from sulphuretted hydrogen, is ready for titrating.
The colour imparted to hot hydrochloric acid solutions by a trace of a ferric compound is so strong, and the reducing action of stannous chloride is so rapid, that a method of titration is based upon the quantity of a standard solution of stannous chloride required to completely decolorise a solution containing ferric iron. This method is more especially adapted for the assay of liquors containing much ferric iron and of those oxidised ores which are completely soluble in hydrochloric acid. It must be remembered, however, that it only measures the ferric iron present, and when (as is generally the case) the total iron is wanted,[Pg 245] it is well to calcine the weighed portion of ore previous to solution in order to get the whole of the iron into the higher state of oxidation, since many ores which are generally supposed to contain only ferric iron carry a considerable percentage of ferrous.
The stannous chloride solution is made by dissolving 20 grams of the commercial salt (SnCl2.2H2O) in 100 c.c. of water with the help of 20 c.c. of dilute hydrochloric acid, and diluting to a litre. The solution may be slightly opalescent, but should show no signs of a precipitate. The strength of this is about equivalent to 1 gram of iron for each 100 c.c. of the solution, but it is apt to lessen on standing, taking up oxygen from the air, forming stannic chloride. A larger proportion of hydrochloric acid than is ordered above would remove the opalescence, but at the same time increase this tendency to atmospheric oxidation, as the following experiments show. The stannous chloride solution (20 c.c.) was mixed with varying amounts of strong hydrochloric acid (sp. g. 1.16), diluted to 100 c.c., and exposed in open beakers for varying lengths of time; and the residual stannous chloride measured by titration with permanganate. The quantities required were as follows:—
| Time Exposed. | 50 per cent. Acid. | 10 per cent. Acid. | 1 per cent. Acid. |
| 1 hour | 33.2 c.c. | 34.4 c.c. | 34.5 c.c. |
| 1 day | 5.0 " | 24.0 " | 27.6 " |
| 2 days | 3.0 " | 14.5 " | 21.3 " |
These indicate very clearly the increased susceptibility to oxidation in strongly acid solutions.
A standard solution of ferric chloride is prepared in the same manner as that described under the experiments on the methods of reduction; but it should be of twice the strength, so that 100 c.c. may contain 1 gram of iron. This solution is used for standardising the stannous chloride when required; and must be carefully prepared; and tested for the presence of nitric acid.
The titration is more limited in its application than either of the oxidising processes because of the restrictions as to bulk, quality and quantity of free acid present, and other conditions of the solution to be assayed. The following experiments show the conditions necessary for a successful titration.
Effect of Varying Temperature.—Twenty c.c. of ferric chloride solution with 20 c.c. of strong hydrochloric acid, diluted to 50 c.c., gave the following results when titrated:—
| Temperature | 15° | 30° | 70° | 100° |
| Stannous chloride required | 22.8 c.c. | 22.0 c.c. | 22.1 c.c. | 22.0 c.c. |
The finishing point, however, is more distinct the hotter the[Pg 246] solution; so that it is best in all cases to run the standard into the boiling solution.
Effect of Varying Bulk.—Solutions containing the same quantity of iron and acid as the last, but diluted to various bulks, and titrated while boiling, gave the following results:—
| Bulk | 30 c.c. | 100 c.c. | 500 c.c. |
| Stannous chloride required | 21.5 " | 21.7 " | 24.3 " |
Effect of Varying Quantities of Hydrochloric Acid.—In these experiments the bulk before titration was 50 c.c. except in the last, in which it was 70 c.c. With less than 5 c.c. of strong hydrochloric acid the finishing point is indistinct and prolonged.
| Strong hydrochloric acid present | 5 c.c. | 10 c.c. | 20 c.c. | 30 c.c. | 50 c.c. |
| Stannous chloride required | 21.1 " | 21.1 " | 21.2 " | 21.8 " | 22.2 " |
Effect of Free Sulphuric Acid.—In these experiments 20 c.c. of hydrochloric acid were present, and the bulk was 50 c.c.
| Strong sulphuric acid present | — c.c. | 3 c.c. | 5 c.c. | 10 c.c. |
| Stannous chloride required | 21.6 " | 22.3 " | 22.9 " | 23.1 " |
This interference of strong sulphuric acid may be completely counteracted by somewhat modifying the mode of working. Another experiment, like the last of this series, required 21.6 c.c.
Effect of Foreign Salts.—Experiments in which 10 grams of various salts were added showed them to be without effect. The results were as follows:—
| Salt present | — | AmCl | Am2SO4 | MgCl2 |
| Stannous chloride required | 21.6 c.c. | 21.6 c.c. | 21.6 c.c. | 21.6 c.c. |
| Salt present | CaCl2 | FeCl2 | Al2Cl6 | |
| Stannous chloride required | 21.8 c.c. | 21.6 c.c. | 21.6 c.c. |
Effect of Varying Iron.—Titrating a solution (with 20 c.c. of hydrochloric acid) measuring 50 c.c., and kept boiling, the quantity of stannous chloride solution required is practically proportional to the iron present.
| Ferric chloride added | 1 c.c. | 10 c.c. | 20 c.c. | 50 c.c. | 100 c.c. |
| Stannous chloride required | 1.1 " | 10.5 " | 20.6 " | 51.4 " | 102.6 " |
The student, having practised some of the above experiments, may proceed to the assay of an iron ore.
Determination of Iron in Brown Iron Ore.—Weigh up 1 gram of the dried and powdered ore, calcine in the cover of a[Pg 247] platinum crucible, and dissolve up in an evaporating dish[69] with 20 c.c. of strong hydrochloric acid. When solution is complete, dilute to 50 c.c. after replacing any acid that may have been evaporated. Boil, and run in the stannous chloride solution until the colour is faintly yellow; boil again, and continue the addition of the stannous chloride solution, stirring continuously until the solution appears colourless. Note the quantity of the stannous chloride solution required. Suppose this to be 59 c.c. Take 60 c.c. of the standard ferric chloride solution, add 20 c.c. of hydrochloric acid, boil and titrate in the same way as before. Suppose this to require 61 c.c. Then as 61 is equivalent to 60 of the iron solution, 59 is equivalent to 58.13.[70] This gives the percentage. It is not necessary to standardise the stannous chloride solution in this way with each sample assayed, the ratio 61: 60 would serve for a whole batch of samples; but the standardising should be repeated at least once each day.
This method is valuable for the determination of small quantities of iron present as impurities in other metals or ores. It is based on the red coloration developed by the action of potassic sulphocyanate on acid solutions of ferric salts.
Standard Ferric Chloride Solution.—Take 1 c.c. of the ferric chloride solution used for standardising the stannous chloride solution, add 2 c.c. of dilute hydrochloric acid, and dilute to 1 litre with water. 1 c.c. = 0.01 milligram.
Solution of Potassic Sulphocyanate.—Dissolve 60 grams of the salt in water, and dilute to a litre. It should be colourless. Use 10 c.c. for each test.
The quantity of the substance to be weighed for the assay should not contain more than a milligram of iron; consequently, if the ore contain more than 0.1 per cent. of that metal, less than a gram of it must be taken.
The method is as follows:—Weigh up 1 gram of the substance and dissolve in a suitable acid; dilute; and add permanganate of potash solution until tinted. Boil for some time and dilute to 100 c.c. Take a couple of Nessler tubes, holding over 100 c.c., but marked at 50 c.c.; label them "1" and "2"; and into each[Pg 248] put 10 c.c. of the potassic sulphocyanate solution and 2 c.c. of dilute hydrochloric acid. The solutions should be colourless. To "1" add 10 c.c. of the assay solution, and dilute to the 50 c.c. mark. To the other add water, but only to within 5 or 10 c.c. of this mark. Now run in the standard ferric chloride solution from a small burette, 1 c.c. at a time, stirring after each addition till the colour is nearly equal to that of the assay (No. 1). At this stage bring the solution to the same level by diluting, and make a further addition of the standard ferric chloride solution till the colours correspond. The amount of iron will be the same in each tube; that in the standard may be known by reading off the volume from the burette and multiplying by 0.01 milligram.
If the 10 c.c. of the assay solution gave a colour requiring more than 5 or 6 c.c. of the standard ferric chloride solution, repeat the determination, taking a smaller proportion.
The effect of varying conditions on the assay will be seen from the following experiments:—
Effect of Varying Temperature.—The effect of increase of temperature is to lessen the colour; in fact, by boiling, the colour can be entirely removed. All assays are best carried out in the cold.
| 1 | c.c. at 15° | would only show | the colour of | 0.75 | c.c. at 45° |
| 2 | " | " | " | 1.75 | " |
| 5 | " | " | " | 4.0 | " |
Effect of Time.—The effect of increase of time is to increase the colour, as will be seen from the following experiments:—
| 2 | c.c. on standing | 10 minutes | became equal to | 2.25 | c.c. |
| 2 | " | 20 | " | 2.75 | " |
| 2 | " | 40 | " | 3.00 | " |
Effect of Free Acid.—If no acid at all be present, the sulphocyanate of potassium solution removes the colour it first produces, so that a certain amount of acid is necessary to develop the colour. The use of a large excess has a tendency to increase the colour produced.
5 c.c. nitric acid (sp. g. 1.4) read 3.7 c.c. instead of 2 c.c. with the dilute acid.
5 c.c. sulphuric acid (sp. g. 1.32) read 2.2 c.c. instead of 2 c.c. with the dilute acid.
5 c.c. hydrochloric acid (sp. g. 1.16) read 2.5 c.c. instead of 2 c.c. with the dilute acid.
Effect of Foreign Metals.—Lead, mercury, cadmium, bismuth, arsenic, tin, antimony, nickel, cobalt, manganese, aluminium, zinc, strontium, barium, calcium, magnesium, sodium, or potassium, when separately present in quantities of from 100 to 200 times the weight of iron present, do not interfere if they have previously[Pg 249] been brought to their highest oxidised condition by boiling with nitric acid or by treating with permanganate. Arsenic and phosphoric acids interfere unless an excess of free hydrochloric or other acid is present. Oxalic acid (but not tartaric acid) in minute quantities destroys the colour. Nitrous acid strikes a red colour with the sulphocyanate of potassium; consequently, when nitric acid has been used in excess, high results may be obtained. Copper and some other metals interfere, so that in most cases it is advisable to concentrate the iron before estimating it. A blank experiment should always be made with the reagents used in order to determine the iron, if any, introduced during the solution, &c., of the substance assayed.
Determination of Iron in Metallic Copper.—This may be most conveniently done during the estimation of the arsenic. The small quantity of white flocculent precipitate which may be observed in the acetic acid solution before titrating, contains the whole of the iron as ferric arsenate. It should be filtered off, dissolved in 10 c.c. of dilute hydrochloric acid, and diluted to 100 c.c.; 10 c.c. of this may be taken for the estimation. For example: 10 grams of copper were taken, and the iron estimated; 3.0 c.c. of standard ferric chloride solution were used, equivalent to 0.03 milligram of iron; this multiplied by 10 (because only 1/10th of the sample was taken) gives 0.3 milligram as the iron in 10 grams of copper. This equals 0.003 per cent.
In a series of experiments with this method working on 10-gram lots of copper, to which known quantities of iron had been added, the following were the results:—
| Iron present | 0.015% | 0.070% | 0.100% | 0.495% |
| Iron found | 0.015" | 0.061" | 0.087" | 0.522" |
When no arsenic is present in the copper, the iron can be separated by fractionally precipitating with sodic carbonate, dissolving in ammonia, and filtering off the ferric hydrate. Coppers generally carry more iron the less arsenic they contain.
Determination of Iron in Metallic Zinc.—Dissolve 1 gram of zinc in 10 c.c. of dilute hydrochloric acid, adding a drop or two of nitric acid towards the end to effect complete solution. Boil, dilute, and tint with the permanganate of potassium solution; boil till colourless, and dilute to 100 c.c. Take 10 c.c. for the determination. Make a blank experiment by boiling 10 c.c. of dilute hydrochloric acid with a drop or two of nitric acid; add a similar quantity of the permanganate of potassium solution, boiling, &c., as before. The quantity of iron in zinc varies from less than 0.005 to more than 2.0 per cent. When 1 gram is taken and[Pg 250] worked as above, each c.c. of ferric chloride solution required indicates 0.01 per cent. of iron.
Determination of Iron in Metallic Tin.—Cover 1 gram of tin with 5 c.c. of hydrochloric acid, add 1 c.c. of nitric acid, and evaporate to dryness. Take up with 2 c.c. of dilute hydrochloric acid, add 10 c.c. of the potassic sulphocyanate solution, and make up to 50 c.c. Probably the colour developed will be brown instead of red owing to the presence of copper; in this case, add to the standard as much copper as the assay is known to contain (which must have previously been determined; see Copper); the titration is then carried out in the usual way.
Or the iron may be separated from the copper in the tin by the following process:—Dissolve 5 grams of metal in 30 c.c. of hydrochloric acid and 5 c.c. of nitric acid, and evaporate to dryness. Take up with 5 c.c. of dilute hydrochloric acid, add 10 grams of potash dissolved in 30 c.c. of water, and warm till the tin is dissolved. Pass sulphuretted hydrogen, boil, cool, and filter. The iron and copper will be in the precipitate. They are separated in the ordinary manner.
1. Calculate from the following determinations the percentages of ferrous, ferric, and total iron in the sample of ore used.
1 gram of ore dissolved and titrated required 26.7 c.c. of bichromate of potassium solution.
1 gram of ore dissolved, reduced, and titrated required 43.5 c.c. of bichromate of potassium solution.
Standard = 1.014.
2. One gram of an ore contained 0.307 gram of ferrous iron and 0.655 gram of total iron. The iron existing as oxide, what are the percentages of ferrous oxide (FeO) and ferric oxide (Fe2O3) in the ore?
3. One gram of brown iron ore dissolved in hydrochloric acid required 59.2 c.c. of stannous chloride (standard = 0.930). Another gram dissolved in acid and titrated with "permanganate" required 8.2 c.c. (standard = 0.4951). Calculate the percentages of ferrous, ferric, and total iron.
4. Another gram of the same ore, roasted, dissolved and titrated with stannous chloride, required 63.5 c.c. To what extent does this result confirm the others?
5. Two grams of a metal were dissolved and diluted to 100 c.c. Five c.c. were taken for a colorimetric determination, and required 4.5 c.c. of the standard ferric chloride solution. What is the percentage of iron in the metal?[Pg 251]
Nickel and cobalt are closely related in their chemical properties, and may best be considered together. Nickel is the commoner of the two, and is met with in commerce alloyed with copper and zinc as German silver; as also in the coinage of the United States and on the Continent. It is used for plating polished iron and steel goods, forming a coating little liable to rust and taking a good polish. The ores of nickel are not very common. Kupfernickel and chloanthite are arsenides of nickel with, generally, more or less iron and cobalt. Noumeite and garnierite are hydrated silicates of nickel and magnesia. The chief sources of nickel are these silicates, which are found in large quantity in New Caledonia; and a pyrites found in Norway, containing three or four per cent. of the metal. In smaller quantities it is more widely distributed, being frequently met with in copper ores; consequently, commercial copper is rarely free from it.
Nickel is readily soluble in moderately concentrated nitric acid. Its salts are mostly green, and soluble in excess of ammonia, forming blue solutions; in these respects it resembles copper. The acid solutions, however, are not precipitated by sulphuretted hydrogen, although in alkaline solutions a black sulphide is formed which is insoluble in dilute hydrochloric acid. If the sulphide is formed in a solution containing much free ammonia, the precipitation is incomplete, some sulphide remaining in the solution and colouring it dark brown. These reactions serve to distinguish and separate nickel from other metals, except cobalt. If the separated sulphide be heated in a borax bead, the colour obtained will be a sherry brown in the outer flame, and grey or colourless in the inner flame if nickel only is present. In the presence of cobalt these colours are masked by the intense and characteristic blue yielded in both flames by that metal.
The dry assay of nickel (cobalt being at the same time determined) is based on the formation of a speise which will carry the cobalt, nickel, copper, and some of the iron of the ore in combination with arsenic. A speise of this kind, fused and exposed at a red heat to air, first loses arsenide of iron by oxidation. It is only when the iron has been oxidised that the arsenide of cobalt begins to be attacked; and when the removal of the cobalt is complete, the nickel commences to pass into the slag, the copper being left till last. The changes are rendered evident by fusion[Pg 252] in contact with borax. The process is as follows:—Weigh up 5 grams of the ore, and calcine thoroughly on a roasting dish in the muffle. Rub up with some anthracite, and re-roast. Mix intimately with from 3 to 5 grams of metallic arsenic, and heat in a small covered clay crucible at dull redness in a muffle until no more fumes of arsenic come off (about 15 minutes). Take out the crucible, and inject a mixture of 20 grams of carbonate of soda, 5 grams of flour, and 2 grams of fused borax. Place in the wind furnace, and raise the temperature gradually until the charge is in a state of tranquil fusion. Pour; when cold, detach the button of speise, and weigh.
Weigh out carefully a portion of about 1 gram of it. Place a shallow clay dish in the muffle, and heat it to bright redness; then add about 1.5 gram of borax glass wrapped in a piece of tissue paper; when this has fused, drop the piece of speise into it. Close the muffle until the speise has melted, which should be almost at once. The arsenide of iron will oxidise first, and when this has ceased the surface of the button brightens. Remove it from the muffle, and quench in water as soon as the button has solidified. The borax should be coloured slightly blue. Weigh: the loss is the arsenide of iron. Repeat the operation with the weighed button on another dish, using rather less borax. Continue the scorification until a film, green when cold, floating on the surface of the button shows that the nickel is beginning to oxidise. Cool, separate, and weigh the button as before. The loss is the arsenide of cobalt.
If copper is absent, the speise is now arsenide of nickel.
The weight of nickel corresponding to the arsenide got is calculated by multiplying by 0.607; and, similarly, the weight of the cobalt is ascertained by multiplying the loss in the last scorification by 0.615.[71] It must be remembered that the nickel and cobalt so obtained are derived from a fraction only of the speise yielded by the ore taken, so that the results must be multiplied by the weight of the whole of the speise, and divided by the weight of the fragment used in the determination. As an example, suppose 5 grams of ore gave 3.3 grams of speise, and 1.1 gram of this gave 0.8 gram of nickel arsenide. Then—
| 0.8×0.607 | = | 0.4856 | gram of nickel |
| 0.4856×3.3/1.1 | = | 1.456 | gram of nickel |
And this being obtained from 5 grams of ore is equivalent to 29.12 per cent.
When copper is also present, weigh up accurately about 0.5 gram of gold, and place it on the scorifier with the button of nickel and copper arsenide, using borax as before. Scorify until[Pg 253] the button shows the bluish-green colour of a fused gold-copper alloy. Then cool, and weigh the button of copper and gold. The increase in weight of the gold button gives the copper as metal. The weight of the copper multiplied by 1.395 is the weight of the copper arsenide (Cu3As) present. The difference will be the nickel arsenide.
The student should enter the weighings in his book as follows:
| Ore taken | — | grams |
| Speise got | — | " |
| Speise taken | — | grams | |
| Arsenides of | cobalt, nickel, and copper | — | " |
| " | nickel and copper | — | " |
| Gold added | — | " | |
| Gold and copper got | — | " | |
| Showing Cobalt | — | per cent. | |
| Nickel | — | " | |
| Copper | — | " |
Solution and Separation.—Two or three grams of a rich ore, or 5 to 10 grams if poor, are taken for the assay. If much arsenic is present (as is usually the case), the ore must be calcined before attacking with acids. Transfer to a flask; and boil, first with hydrochloric acid until the oxides are dissolved, and then with the help of nitric acid, until nothing metalliferous is left. Dilute, nearly neutralise with soda, and separate the iron as basic acetate,[72] as described in page 233. Through the filtrate pass sulphuretted hydrogen till saturated. Allow to settle (best overnight), filter, and wash. Transfer the precipitate to a beaker, and dissolve in nitric acid. Dilute with water, pass sulphuretted hydrogen, and filter off the precipitate, if any. Boil off the gas, add ammonia until a precipitate is formed, and then acidify somewhat strongly with acetic acid. Pass sulphuretted hydrogen in a slow stream until any white precipitate of zinc sulphide, there may be, begins to darken. Filter; to the filtrate add ammonia, and pass sulphuretted hydrogen. The precipitate will contain the nickel and cobalt as sulphides.
Where small quantities of nickel and cobalt are present, and an approximate determination is sufficient, they can be concentrated as follows:—Remove the copper, &c., by passing sulphuretted hydrogen through the acid solution and filtering; add ammonia[Pg 254] to the filtrate, and again pass sulphuretted hydrogen; then heat nearly to boiling, and filter. Dissolve the precipitate off the filter with dilute hydrochloric acid; the residue will contain nearly all the nickel and cobalt as sulphides.
Separation of Nickel and Cobalt.—Dissolve the sulphides separated as above in nitric acid; render alkaline with a solution of potash, then acidify with acetic acid; add a concentrated solution of nitrite of potash. The liquid after this addition must have an acid reaction. Allow to stand for 24 hours in a warm place. Filter off the yellow precipitate of nitrite of potash and cobalt, and wash with a 10 per cent. solution of acetate of potash. The cobalt is determined in the precipitate in the way described under Cobalt. The nickel is separated from the solution by boiling with sodic hydrate, filtering, and dissolving the precipitate in nitric acid. The solution will contain the nickel.
The solution, which contains the nickel free from other metals, is heated, and a solution of sodic hydrate added in slight excess. The precipitate is filtered off, washed with boiling water, dried, ignited at a red heat, and weighed when cold. The ignited substance is nickel oxide (NiO), and contains 78.67 per cent. of nickel. The oxide is a green powder, readily and completely soluble in hydrochloric acid, and without action on litmus paper. It is very easily reduced by ignition in hydrogen to metallic nickel.
Nickel is also determined by electrolysis, as follows:—The nitric acid solution is rendered strongly ammoniacal, and placed under the electrolytic apparatus used for the copper assay. Three cells (fig. 56), however, must be used, coupled up for intensity, that is, with the zinc of one connected with the copper of the next. The electrolysis is allowed to go on overnight, and in the morning the nickel will be deposited as a bright and coherent film. A portion of the solution is drawn off with a pipette; if it smells of ammonia, has no blue colour, and gives no precipitate with ammonic sulphide, the separation is complete. Wash the cylinder containing the deposited metal, first with water and then with alcohol, as in the copper assay. Dry in the water oven, and weigh. The increase in weight is metallic nickel.[Pg 255]
As an example:—There was taken 1 gram of a nickel alloy used for coinage. It was dissolved in 10 c.c. of nitric acid, and diluted to 100 c.c. with water. The copper was then precipitated by electrolysis. It weighed 0.734 gram. The solution, after electrolysis, was treated with sulphuretted hydrogen, and the remaining copper was thrown down as sulphide, and estimated colorimetrically. This amounted to 3-1/2 milligrams. The filtrate was evaporated, treated with ammonia, warmed, and filtered. The ferric hydrate was dissolved in dilute acid, and reprecipitated, dried, ignited, and weighed. Its weight was 0.0310 gram. The two filtrates were mixed, and reduced in bulk to about 50 c.c.; a considerable excess of ammonia was added, and the nickel precipitated by electrolysis. It weighed 0.2434 gram. These quantities are equivalent to:
| Copper | 73.75 | per cent. |
| Nickel | 24.34 | " |
| Iron | 2.17 | " |
| ——— | ||
| 100.26 |
An alkaline solution of potassium cyanide, to which a little potassium iodide has been added, can be assayed for its strength in cyanide by titrating with a standard solution of silver nitrate. Nickel interferes with this assay, doing the work of its equivalent of silver; and the quantity of nickel present can be calculated from the amount of its interference in the titration. A volumetric assay for nickel is based on this. It has the disadvantage of all indirect titrations in that it requires two standard solutions. On the other hand it gives good results even under unfavourable conditions, and is applicable in the presence of much zinc. Small quantities of cobalt will count as so much nickel, but larger quantities make the assay unworkable. Some of the other metals—lead for example—have no appreciable effect; but practically the solution demands a preliminary treatment which would result in their removal. Nevertheless it is a very satisfactory method and makes the determination of nickel quick and comparatively easy in most cases.
The standard solution of silver nitrate is made by dissolving 14.48 grams of recrystallised silver nitrate in distilled water and diluting to 1 litre: 100 c.c. of this solution are equivalent to 0.25 gram of nickel.[73]
The standard solution of potassium cyanide should be made so as to be exactly equal to the silver nitrate solution. This can be done as follows: Weigh up 12 grams of good potassium cyanide (95 per cent.), dissolve in water, add 50 c.c. of a 10 per cent. solution of sodium hydrate and dilute to 1 litre. Fill one burette with this and another with the solution of silver nitrate. Run 50 c.c. of the cyanide into a flask; add a few drops of potassium iodide solution and titrate with the standard silver nitrate until there is a distinct permanent yellowish turbidity. The titration is more fully described under Cyanide, p. 165. The cyanide solution will be found rather stronger than the silver nitrate; dilute it so as to get the two solutions of equal value. For example, 51.3 c.c. of silver nitrate may have been required: then add 1.3 c.c. of water to each 50 c.c. of the cyanide solution remaining. If the full 950 c.c. are available, then add to them 24.7 c.c. of water. After mixing, take another 50 c.c. and titrate with the silver nitrate; the two solutions should now be exactly equal. The cyanide solution, being strongly alkaline with soda, keeps very well; but its strength should be checked from time to time by titrating with silver nitrate; should there be any slight inequality in the strengths of the two solutions it is easily allowed for in the calculations.
The titration.—The solution, containing not much more than 0.1 gram of nickel, and free from the interfering metals, must be cooled. It is next neutralised and then made strongly alkaline with a solution of soda (NaHO); an excess of 20 or 30 c.c. suffices. This will produce a precipitate. The cyanide solution is now run in from a burette until the solution clears, after which an excess of about 20 c.c. is added. It is well to use some round number of c.c. to simplify the calculation. Add a few drops of potassium iodide solution, and run in the standard solution of silver nitrate from a burette. This should be done a little at a time, though somewhat rapidly, and with constant shaking, till a permanent yellow precipitate appears. If the addition of the cyanide did not result in a perfectly clear solution, this is because something besides nickel is present. The residue may be filtered off, though with a little practice the finishing-point may be detected with certainty in the presence of a small precipitate. If the student has the slightest doubt about a finish he should run in another 5 c.c. of the cyanide and again finish with silver nitrate. The second result will be the same as the first. For example, if 40 c.c. of cyanide and 30 c.c. of silver nitrate were required at the first titration, then the 45 c.c. of cyanide in the second titration will require 35 c.c. of silver nitrate. The difference between the quantities[Pg 257] of the two solutions used in each case will be 10 c.c. It is this difference in the readings of the two burettes which measures the quantity of nickel present. Each c.c. of the difference is equal to .0025 gram of nickel. But if the cyanide solution is not exactly equal in strength to the silver nitrate, the quantity of cyanide used should be calculated to its equivalent in silver nitrate before making the subtraction.
The following experimental results illustrate the accuracy of the assay and the effect upon it of varying conditions. A solution containing 1 gram of nickel sulphate (NiSO4.6H2O) in 100 c.c. was used. By a separate assay the sulphate was found to contain 22.25 per cent. of nickel. For the sake of simplicity the results of the experiments are stated in weights of nickel in grams.
Effect of varying excess of Cyanide Solution.—In each experiment there was 20 c.c. of the nickel solution, equal to .0445 gram of nickel. There were also 10 c.c. of soda solution, 3 or 4 drops of potassium iodide and sufficient water to bring the bulk to 100 c.c. before titrating.
| Cyanide in excess | 6 c.c. | 4 c.c. | 8 c.c. | 12 c.c. | 25 c.c. |
| Nickel found | .0434 | .0436 | .0440 | .0442 | .0444 |
Although the difference between the highest and lowest of these results is only 1 milligram, their meaning is quite obvious. The excess of cyanide should not be less than 20 c.c.
Effect of varying the quantity of Soda.—There were two series of experiments, one with 2 c.c. of nickel solution (= .0044 gram of nickel), the other with 20 c.c. The conditions were as before, except that the quantity of soda was varied.
| Soda added | 5 c.c. | 15 c.c. | 30 c.c. | |
| Nickel found, | 1st series | .0037 | .0042 | .0045 |
| " " | 2nd series | .0444 | .0444 | .0442 |
These show that the presence of much soda, though it has only a small effect, is beneficial rather than otherwise. Ammonia has a bad effect, if present in anything like the same quantities.
Effect of varying the Nickel.—In experiments with 10, 20, and 40 c.c. of the nickel solution, the results were:—
| Nickel present | .0222 | .0445 | .0890 |
| Nickel found | .0220 | .0442 | .0884 |
Effect of Zinc.—In these experiments 20 c.c. of nickel solution (= .0445 gram of nickel), 10 c.c. of soda, 6 drops of potassium iodide and water to 100 c.c. were used. The excess of cyanide was purposely kept at from 10 to 15 c.c., which is hardly sufficient.
| Zinc added | 0 | .25 gram. | .5 gram. |
| Nickel found | .0442 | .0440 | .0407 |
On increasing the excess of cyanide to over 20 c.c. and doubling the quantity of soda, the experiment with 0.5 gram of zinc gave 0.441 gram of nickel. Hence the titration is satisfactory in the presence of zinc provided that not fewer than 20 or 30 c.c. of soda are used, and that the excess of cyanide is such that not fewer than 20 or 30 c.c. of silver nitrate are required in the titration. Moreover, these precautions should be taken whether zinc is present or not.
Effect of other Metals.—If metals of the first and second groups are present they should be removed by passing sulphuretted hydrogen and filtering. If iron is present it must be removed, since ferrous salts use up much cyanide, forming ferrocyanides, and ferric salts yield ferric hydrate, which obscures the end reaction. Hence the sulphuretted hydrogen must be boiled off and the iron removed as basic ferric acetate by the method described on p. 233. If the precipitate is bulky it should be dissolved in a little dilute acid, neutralised and again precipitated as basic acetate. The nickel will be in the two filtrates. In the absence of manganese and cobalt the titration may be made without further separation.
Manganese does not directly interfere, but the precipitated hydrate, which rapidly darkens through atmospheric oxidation, obscures the end reaction. It may be removed by passing sulphuretted hydrogen through the filtrate from the acetate separation: sulphides of nickel, cobalt and zinc will be precipitated, whilst manganese remains in solution: the addition of more sodium acetate may assist the precipitation. The precipitate must be filtered off and dissolved in nitric acid: the solution should be evaporated to dryness. The filtrate may retain a little nickel; if so, add ammonia till alkaline, then acidify with acetic acid and again filter; any small precipitate obtained here should be added to that first obtained.
It is only when cobalt is present that any further separation is required. Cobalt hydrate takes up oxygen from the air, and on adding potassium cyanide some may refuse to dissolve; and the solution itself acquires a brown colour, which becomes deeper on standing. At this stage the cobalt is easily separated. The solution containing the nickel and cobalt with no great excess of acid, is made alkaline by adding 20 c.c. of soda exactly as in preparing for a titration. So, too, the solution of cyanide is added so as to have an excess of 20 or 30 c.c.; the solution may have a brown colour, but if it is not quite clear it must be filtered. Then warm (boiling is not needed) and add from 50 to 100 c.c. of bromine water. This throws down all the nickel as black peroxide[Pg 259] in a condition easy to filter. Filter it off and wash with water. The precipitate can be dissolved off the filter with the greatest ease by a little warm sulphurous acid. The filtrate and washings, boiled till free from sulphurous acid, yield the nickel as sulphate in a clean condition.
Determination of Nickel in Nickel Sulphate Crystals.—Take 0.5 gram of the salt, dissolve in 50 c.c. of water and add 25 c.c. of solution of soda. Run in from a burette, say, 60 c.c. "cyanide." Add a few drops of potassium iodide and titrate back with "silver nitrate." Suppose 15.5 c.c. of the latter is required. Then 15.5 c.c. subtracted from 60 c.c. leaves 44.5 c.c., and since 100 c.c. = 0.25 gram of nickel, 44.5 c.c. will equal 0.11125 gram of nickel. This in 0.5 gram of the salt equals 22.25 per cent.
Determination of Nickel in German Silver.—Weigh up 0.5 gram of the alloy, and dissolve in a dish with 5 or 10 c.c. of dilute nitric acid. Add 5 c.c. of dilute sulphuric acid and evaporate till all the nitric acid is removed. Cool, take up with 50 c.c. of water, and when dissolved pass sulphuretted hydrogen through the solution. Filter off the precipitate and wash with water containing sulphuretted hydrogen and dilute sulphuric acid. Boil down the filtrate and washings to get rid of the excess of the gas; add some nitric acid and continue the boiling. Cool, neutralise the excess of acid with soda, add 1 gram of sodium acetate and boil. Filter off the precipitate which contains the iron. The filtrate, cooled and rendered alkaline with soda, is ready for the titration.
Occurs less abundantly than nickel. Its chief ores are smaltite and cobaltite, which are arsenides of cobalt, with more or less iron, nickel, and copper. It also occurs as arseniate in erythrine, and as oxide in asbolan or earthy cobalt, which is essentially a wad carrying cobalt.
It is mainly used in the manufacture of smalts for imparting a blue colour to glass and enamels. The oxide of cobalt forms coloured compounds with many other metallic oxides. With oxide of zinc it forms "Rinman's green"; with aluminia, a blue; with magnesia, a pink. This property is taken advantage of in the detection of substances before the blow-pipe.
The compounds of cobalt in most of their properties closely resemble those of nickel, and the remarks as to solution and separation given for the latter metal apply here. Solutions of cobalt are pink, whilst those of nickel are green.[Pg 260]
The detection of cobalt, even in very small quantity, is rendered easy by the strong blue colour which it gives to the borax bead, both in the oxidising and in the reducing flame. It is concentrated from the ore in the same way as nickel, and should be separated from that metal by means of potassic nitrite in the way described. The dry assay of cobalt has been given under Nickel.
The yellow precipitate from the potassium nitrite, after being washed with the acetate of potash, is washed with alcohol, dried, transferred to a weighed porcelain crucible, and cautiously ignited with an excess of strong sulphuric acid. The heat must not be sufficient to decompose the sulphate of cobalt, which decomposition is indicated by a blackening of the substance at the edges. The salt bears a low red heat without breaking up. If blackening has occurred, moisten with sulphuric acid, and ignite again. Cool and weigh. The substance is a mixture of the sulphates of cobalt and potash (2CoSO4 + 3K2SO4), and contains 14.17 per cent. of cobalt.
Cobalt is also gravimetrically determined, like nickel, by electrolysis, or by precipitation with sodic hydrate. In the latter case, the ignited oxide will be somewhat uncertain in composition, owing to its containing an excess of oxygen. Consequently, it is better to reduce it by igniting at a red heat in a current of hydrogen and to weigh it as metallic cobalt.
1. In the dry assay of an ore containing cobalt, nickel, and copper, the following results were obtained. Calculate the percentages. Ore taken, 5 grams. Speise formed, 0.99 gram. Speise taken. 0.99 gram. Arsenides of cobalt, nickel, and copper got, 0.75 gram. Arsenide of nickel and copper got, 0.54 gram. Gold added, 0.5 gram. Gold and copper got, 0.61 gram.
2. Calculate the percentage composition of the following compounds: Co2As, Ni2As, and Cu2As.
3. A sample of mispickel contains 7 per cent. cobalt. What weight of the mixed sulphates of potash and cobalt will be obtained in a gravimetric determination on 1 gram of the ore?
4. 0.3157 gram of metal was deposited by the electrolysis of a nickel and cobalt solution. On dissolving in nitric acid and determining the cobalt 0.2563 gram of potassium and cobalt sulphates were got. Find the weights of cobalt and nickel present in the deposit.
5. What should be the percentage composition of pure cobaltite, its formula being CoAsS?[Pg 261]
Zinc occurs in nature most commonly as sulphide (blende); it also occurs as carbonate (calamine) and silicate (smithsonite). Each of these is sufficiently abundant to be a source of the metal.
The metal is known in commerce as "spelter" when in ingots, and as sheet zinc when rolled. It is chiefly used in the form of alloys with copper, which are known as brasses. It is also used in the form of a thin film, to protect iron goods from rusting—galvanised iron.
Ores of zinc, more especially blende, are met with in most lead, copper, gold, and silver mines, in larger or small quantities scattered through the lodes. Those ores which generally come under the notice of the assayer are fairly rich in zinc; but alloys and metallurgical products contain it in very varying proportions.
Zinc itself is readily soluble in dilute acids; any residue which is left after boiling with dilute hydrochloric or sulphuric acid consists simply of the impurities of the metal; this is generally lead.
All zinc compounds are either soluble in, or are decomposed by, boiling with acids, the zinc going into solution. Zinc forms only one series of salts, and these are colourless. Their chief characteristic is solubility in an alkaline solution, from which sulphuretted hydrogen produces a white precipitate of zinc sulphide. Zinc is detected by dissolving the substance in hydrochloric or nitric acid, boiling, and adding sodic hydrate in excess, filtering, and adding ammonic sulphide to the filtrate. The precipitate contains the zinc, which can be dissolved out by boiling with dilute sulphuric acid, and detected by the formation of a white precipitate on the addition of potassic ferrocyanide.
The dry assay of zinc can only be made indirectly, and is unsatisfactory. Zinc is volatile, and at the temperature of its reduction is a gas. It is impracticable to condense the vapour so as to weigh the metal, consequently its amount is determined by loss. The following method gives approximate results: Take 10 grams of the dried and powdered ore and roast, first at a low temperature and afterwards at a higher one, with the help of carbonate of ammonia to decompose the sulphates formed; cool and weigh. The metals will be present as oxides. Mix with 2 grams of powdered charcoal and charge into a black-lead crucible heated to whiteness, cover loosely, and leave in the furnace for about a quarter of an hour. Uncover and calcine the residue, cool and weigh. The loss in weight multiplied by 8.03 gives the percentage of zinc in the ore.[Pg 262]
Solution and separation may be effected as follows: Treat 1 or 3 grams of the substance with 10 or 30 c.c. of hydrochloric acid or aqua regia; evaporate to dryness; take up with 10 c.c. of hydrochloric acid and dilute to 100 c.c.; heat nearly to boiling; saturate with sulphuretted hydrogen; filter, and wash with water acidulated with hydrochloric acid. Boil off the sulphuretted hydrogen and peroxidise with a few drops of nitric acid. Cool; add caustic soda till nearly, but not quite, neutralised, and separate the iron as basic acetate by the method described under Iron. To the filtrate add ammonia till alkaline, and pass sulphuretted hydrogen. Allow to settle and decant on to a filter. Dissolve off the precipitate from the filter with hot dilute hydrochloric acid. The solution will contain the zinc, together with any manganese the ore contained, and, perhaps, traces of nickel and cobalt. If the zinc is to be determined volumetrically, and manganese is present, this latter is separated with carbonate of ammonia, as described further on; but if a gravimetric method is used, and only small quantities of manganese are present, it is better to proceed as if it were absent, and to subsequently determine its amount, which should be deducted.
The solution containing the zinc is contained in an evaporating dish, and freed from sulphuretted hydrogen by boiling, and, if necessary, from an excess of acid by evaporation. The evaporating dish must be a large one. Cautiously add sodium carbonate to the hot, moderately dilute solution, until the liquid is distinctly alkaline, and boil. Allow the precipitate to settle, decant on to a filter, and wash with hot water. Dry, transfer to a porcelain crucible (cleaning the paper as much as possible), add the ash, ignite, and weigh. The substance weighed is oxide of zinc, which contains 80.26 per cent. of the metal. It is a white powder, becoming yellow when heated. It must not show an alkaline reaction when moistened. If it contains manganese this metal will be present as sesquioxide (Mn2O3). Its amount can be determined by dissolving in dilute acid and boiling with an excess of sodic hydrate. The oxide of manganese will be precipitated, and can be ignited and weighed. Its weight multiplied by 1.035 must be deducted from the weight of oxide of zinc previously obtained. The results yielded by the gravimetric determination are likely to be high, since the basic carbonate of[Pg 263] zinc frequently carries down with it more or less soda which is difficult to wash off.
This method is based on the facts that zinc salts in an acid solution decompose potassium ferrocyanide, forming a white insoluble zinc compound; and that an excess of the ferrocyanide can be detected by the brown coloration it strikes with uranium acetate. The method resembles in its working the bichromate iron assay. The standard solution of potassium ferrocyanide is run into a hot hydrochloric acid solution of the zinc until a drop of the latter brought in contact with a drop of the indicator (uranium acetate) on a white plate strikes a brown colour. The quantity of zinc in the solution must be approximately known; run in a little less of the ferrocyanide than is expected will be necessary; test a drop or two of the assay, and then run in, one or two c.c. at a time, until the brown colour is obtained. Add 5 c.c. of a standard zinc solution, equivalent in strength to the standard "ferrocyanide," re-titrate, and finish off cautiously. Of course 5 c.c. must be deducted from the reading on the burette. The precipitate of zinc ferrocyanide formed in the assay solution is white; but if traces of iron are present, it becomes bluish. If the quantity of ferrocyanide required is known within a few c.c., the finishing point is exactly determined in the first titration without any addition of the standard zinc solution. Unfortunately this titration serves simply to replace the gravimetric determination, and does not, as many volumetric processes do, lessen the necessity for a complete separation of any other metals which are present. Most metals give precipitates with ferrocyanide of potassium in acid solutions. If the conditions are held to, the titration is a fairly good one, and differences in the results of an assay will be due to error in the separation. Ferric hydrate precipitated in a fairly strong solution of zinc will carry with it perceptible quantities of that metal. Similarly, large quantities of copper precipitated as sulphide by means of sulphuretted hydrogen will carry zinc with it, except under certain nicely drawn conditions. When much copper is present it is best separated in a nitric acid solution by electrolysis. The titration of the zinc takes less time, and, with ordinary working, is more trustworthy than the gravimetric method.
The standard ferrocyanide solution is made by dissolving 43.2 grams of potassium ferrocyanide (K4FeCy6.3H2O) in water, and diluting to a litre. One hundred c.c. are equal to 1 gram of zinc.[Pg 264]
The standard zinc solution is made by dissolving 10 grams of pure zinc in 50 c.c. of hydrochloric acid and 100 or 200 c.c. of water, and diluting to 1 litre, or by dissolving 44.15 grams of zinc sulphate (ZnSO4.7H2O) in water with 30 c.c. of hydrochloric acid, and diluting to 1 litre. One hundred c.c. will contain 1 gram of zinc.
The uranium acetate solution is made by dissolving 0.2 gram of the salt in 100 c.c. of water.
To standardise the "ferrocyanide" measure off 50 c.c. of the standard zinc solution into a 10 oz. beaker, dilute to 100 c.c., and heat to about 50° C. (not to boiling). Run in 47 or 48 c.c. of the "ferrocyanide" solution from an ordinary burette, and finish off cautiously. Fifty divided by the quantity of "ferrocyanide" solution required gives the standard.
In assaying ores, &c., take such quantity as shall contain from 0.1 to 1 gram of zinc, separate the zinc as sulphide, as already directed. Dissolve the sulphide off the filter with hot dilute hydrochloric acid, which is best done by a stream from a wash bottle. Evaporate the filtrate to a paste, add 5 c.c. of dilute hydrochloric acid, dilute to 100 c.c. or 150 c.c., heat to about 50° C., and titrate. Manganese, if present, counts as so much zinc, and must be specially separated, since it is not removed by the method already given. The following method will effect its removal. To the hydrochloric acid solution of the zinc and manganese add sodium acetate in large excess and pass sulphuretted hydrogen freely. Allow to settle, filter off the zinc sulphide and wash with sulphuretted hydrogen water. The precipitate, freed from manganese, is then dissolved in hydrochloric acid and titrated.
The following experiments show the effect of variation in the conditions of the assay:—
Effect of Varying Temperature.—Using 20 c.c. of the standard zinc solution, 5 c.c. of dilute hydrochloric acid, and diluting to 100 c.c.
| Temperature | 15° C. | 30° C. | 70° C. | 100° C. |
| "Ferrocyanide" required | 20.6 c.c. | 20.3 c.c. | 20.3 c.c. | 20.3 c.c. |
The solution can be heated to boiling before titrating without interfering with the result; but it is more convenient to work with the solution at about 50° C. Cold solutions must not be used.
Effect of Varying Bulk.—These were all titrated at about 50° C., and were like the last, but with varying bulk.
| Bulk | 25.0 | c.c. | 50.0 | c.c. | 100.0 | c.c. | 200.0 | c.c. |
| "Ferrocyanide" required | 20.2 | " | 20.4 | " | 20.3 | " | 20.4 | " |
Any ordinary variation in bulk has no effect.[Pg 265]
Effect of Varying Hydrochloric Acid.— With 100 c.c. bulk and varying dilute hydrochloric acid the results were:—
| Acid added | 0.0 | c.c. | 1.0 | c.c. | 5.0 | c.c. | 10.0 | c.c. | 20.0 | c.c. |
| "Ferrocyanide" required | 24.4 | " | 20.2 | " | 20.3 | " | 20.3 | " | 20.7 | " |
Effect of Foreign Salts.—The experiments were carried out under the same conditions as the others. Five grams each of the following salts were added:—
| Salt added | Ammonic chloride. | Ammonic sulphate. | Sodium chloride. | Sodium sulphate. |
| "Ferrocyanide" required | 20.3 c.c. | 20.5 c.c. | 20.6 c.c. | 20.4 c.c. |
| Salt added | Potassium Nitrate. | Magnesium sulphate. | Nil. | |
| "Ferrocyanide" required | 20.2 c.c. | 20.4 c.c. | 20.4 c.c. |
In a series of experiments in which foreign metals were present to the extent of 0.050 gram in each, with 20 c.c. of zinc solution and 5 c.c. of dilute hydrochloric acid, those in which copper sulphate, ferrous sulphate, and ferric chloride were used, gave (as might be expected) so strongly coloured precipitates that the end reaction could not be recognised. The other results were:—
| "Ferrocyanide" required. | ||||
| With | nothing added. | 20.3 c.c. | ||
| " | 0.050 | gram | lead (as chloride) | 20.9 " |
| " | 0.050 | " | manganese (as sulphate) | 25.5 " |
| " | 0.050 | " | cadmium (as sulphate) | 23.5 " |
| " | 0.050 | " | nickel (as sulphate) | 26.2 " |
Effect of Varying Zinc.—These were titrated under the usual conditions, and gave the following results:—
| Zinc added | 1.0 c.c. | 10.0 c.c. | 20.0 c.c. | 50.0 c.c. | 100.0 c.c. |
| "Ferrocyanide" required | 1.1 " | 10.2 " | 20.3 " | 50.6 " | 101.0 " |
Determination of Zinc in a Sample of Brass.—Take the solution from which the copper has been separated by electrolysis and pass sulphuretted hydrogen until the remaining traces of copper and the lead are precipitated, filter, boil the solution free from sulphuretted hydrogen, put in a piece of litmus paper, and add sodic hydrate solution in slight excess; add 10 c.c. of dilute hydrochloric acid (which should render the solution acid and clear); warm, and titrate.
A sample of 0.5 gram of brass treated in this manner required 16.4 c.c. of "ferrocyanide" (standard 100 c.c. = 0.9909 zinc), which equals 0.1625 gram of zinc or 32.5 per cent.[Pg 266]
Determination of Zinc in Blende.—Dissolve 1 gram of the dried and powdered sample in 25 c.c. of nitric acid with the help of two or three grams of potassium chlorate dissolved in the acid. Evaporate to complete dryness, taking care to avoid spirting. Add 7 grams of powdered ammonium chloride, 15 c.c. of strong ammonia and 25 c.c. of boiling water; boil for one minute and see that the residue is all softened. Filter through a small filter, and wash thoroughly with small quantities of a hot one per cent. solution of ammonium chloride. Add 25 c.c. of hydrochloric acid to the filtrate. Place in the solution some clean lead foil, say 10 or 20 square inches. Boil gently until the solution has been colourless for three or four minutes. Filter, wash with a little hot water; and titrate with standard ferrocyanide.
Determination of Zinc in Silver Precipitate.—This precipitate contains lead sulphate, silver, copper, iron, zinc, lime, &c. Weigh up 5 grams of the sample, and extract with 30 c.c. of dilute sulphuric acid with the aid of heat. Separate the copper with sulphuretted hydrogen, peroxidise the iron with a drop or two of nitric acid, and separate as acetate. Render the filtrate ammoniacal, pass sulphuretted hydrogen; warm, and filter. Dissolve the precipitated zinc sulphide in dilute hydrochloric acid, evaporate, dilute, and titrate. Silver precipitates carry about 2.5 per cent. of zinc.
Metallic zinc is readily soluble in dilute hydrochloric or sulphuric acid, hydrogen being at the same time evolved.[74] The volume of the hydrogen evolved is obviously a measure of the amount of zinc present in the metallic state. The speed with which the reaction goes on (even in the cold) and the insolubility of hydrogen renders this method of assay a convenient one. It is especially applicable to the determination of the proportion of zinc in zinc dust. The apparatus described in the chapter on gasometric method is used. The method of working is as follows: Fill the two burettes with cold water to a little above the zero mark, place in the bottle about 0.25 gram of the substance to be determined, and in the inner phial or test tube 5 c.c. of dilute sulphuric acid; cork the apparatus tightly and allow to stand for a few minutes; then bring the water to the same level in the two burettes by running out through the clip at the bottom. Read off the level of the liquid in the graduated burette. Turn the[Pg 267] bottle over sufficiently to spill the acid over the zinc, and then run water out of the apparatus so as to keep the liquid in the two burettes at the same level, taking care not to run it out more quickly than the hydrogen is being generated. When the volume of gas ceases to increase, read off the level of the liquid, deduct the reading which was started with; the difference gives the volume of hydrogen evolved. At the same time read off the volume of air in the "volume corrector," which must be fixed alongside the gas burettes. Make the correction. For example: A piece of zinc weighing 0.2835 gram was found to give 99.9 c.c. of gas at a time when the corrector read 104 c.c.[75] Then the corrected volume is
104 : 100 :: 99.9 : x.
x = 96.0 c.c.
100 c.c. of hydrogen at 0° C. and 760 mm. is equivalent to 0.2912 gram of zinc; therefore the quantity of zinc found is
100 : 96 :: 0.2912 : x.
x = 0.2795 gram of zinc.
This being contained in 0.2835 gram of metal is equivalent to 98.5 per cent.
As an example of a determination in which reducing the volume of liberated hydrogen to 0° C. and 760 mm. is avoided, the following may be taken:—
0.2315 gram of pure zinc gave 82.1 c.c. of gas;
and the volume of air in the corrector was 103.6 c.c.
0.2835 gram of the assay gave 99.9 c.c. of gas;
and the volume of air in the corrector was 104.0 c.c.;
104 : 103.6 :: 99.9 : x.
x = 99.5 c.c.
This is the volume of gas got in the assay if measured under the same conditions as the standard,
82.1 : 99.5 :: 0.2315 : x.
x = 0.2806.
Then 0.2835 : 0.2806 :: 100: x.
x = 98.9 per cent.
As these assays can be made quickly, it is well for the sake of greater accuracy to make them in duplicate, and to take the mean of the readings. One set of standardisings will do for any number of assays. The student must carefully avoid unnecessary handling of the bottle in which the zinc is dissolved.
Colorimetric Method.—Zinc salts being colourless, there is no colorimetric determination.[Pg 268]
Take 20 grams of zinc, and dissolve them in dilute nitric acid; boil, allow to settle; filter; wash, dry; ignite the precipitate, if any, and weigh as oxide of tin. Examine this for arsenic.
Lead.—Add ammonia and carbonate of ammonia to the liquid, and boil, filter off the precipitate, wash with hot water. Digest the precipitate with dilute sulphuric acid; filter, wash, and weigh the sulphate of lead.
Iron.—To the filtrate from the sulphate of lead add ammonia, and pass sulphuretted hydrogen; digest, and filter. (Save the filtrate.) Dissolve the precipitate in hydrochloric acid, oxidise with nitric acid, and precipitate with ammonia. Wash, ignite, and weigh as ferric oxide. Calculate to iron.
Arsenic.—To the filtrate from the sulphide of iron add hydrochloric acid in slight excess; filter off, and wash the precipitate. Rinse it back into the beaker, dissolve in nitric acid, filter from the sulphur, and add ammonia, in excess, and magnesia mixture. Filter off the ammonic-magnesic arsenate, and wash with dilute ammonia. Dry, ignite with nitric acid, and weigh as magnesic pyrarsenate. Calculate to arsenic, and add to that found with the tin.
Copper.—To the filtrate from the ammonia and ammonic carbonate add sulphuric acid in small excess, and pass sulphuretted hydrogen. Allow to settle, filter, and wash. Rinse the precipitate into a beaker, boil with dilute sulphuric acid, and filter. (Save the filtrate.) Dry, burn the paper with the precipitate, treat with a drop or two of nitric acid, ignite, and weigh as copper oxide. Calculate to copper.
Cadmium.—To the filtrate from the sulphide of copper add ammonia, so as to nearly neutralise the excess of acid, and pass sulphuretted hydrogen. Collect and weigh the precipitate as cadmium sulphide, as described under Cadmium.
1. What weight of hydrogen will be evolved in dissolving 1 gram of zinc in dilute sulphuric acid?
2. How many c.c. would this quantity of hydrogen measure at 0° C. and 760 m.m.? (1 litre weighs 0.0896 gram).
3. 0.23 gram of zinc are found to give 77.9 c.c. of hydrogen. In another experiment under the same conditions 80.2 c.c. are got. What weight of zinc was used for the second experiment?[Pg 269]
4. A sample of blende is found to contain 55 per cent. of zinc. What percentage of zinc sulphide did the sample contain?
5. How much metallic lead would be precipitated from a solution of lead acetate by 1 gram of zinc?
Cadmium occurs in nature as cadmium sulphide in greenockite, CdS, which is very rare. It is widely diffused in calamine, blende, and other zinc ores, forming, in some cases, as much as 2 or 3 per cent. of the ore. Oxide of cadmium forms the "brown blaze" of the zinc smelters.
Sulphide of cadmium is used as a pigment (cadmium yellow); and the metal and some of its salts are useful reagents.
The salts of cadmium closely resemble those of zinc. The hydrate, however, is insoluble in excess of potash, and the sulphide is insoluble in dilute acids. It forms only one series of salts.
Cadmium is detected by giving with sulphuretted hydrogen in solutions, not too strongly acid, a yellow precipitate, which is insoluble in solutions of the alkalies, alkaline sulphides, or cyanide of potassium.
Solution and Separation.—Substances containing cadmium are soluble in acids. The solution is evaporated to dryness (to render any silica that may be present insoluble) and taken up with 10 c.c. of dilute hydrochloric acid. Dilute to 100 c.c., and pass sulphuretted hydrogen. Filter, digest the precipitate with soda, wash, and boil with dilute sulphuric acid. Filter; the filtrate contains the cadmium and, possibly, a small quantity of zinc, from which it is best separated by reprecipitating with sulphuretted hydrogen.
The solution containing the cadmium freed from the other metals is precipitated with sulphuretted hydrogen in a moderately-acid solution. The precipitate is collected on a weighed filter, and washed, first with an acid solution of sulphuretted hydrogen, and afterwards with water. It is dried at 100° C. and weighed. If free sulphur is suspected to be present, extract with bisulphide of carbon, and again weigh. The residue is cadmium sulphide, which contains 77.78 per cent. of cadmium. It is a yellow powder insoluble in solutions of the alkalies, alkaline sulphides, or cyanide of potassium. It dissolves readily in acid. It cannot be ignited in a current of hydrogen without loss.[Pg 270]
The solution containing the cadmium is concentrated by evaporation, and mixed with an excess of oxalic acid and alcohol. The precipitate is filtered, washed with alcohol, dissolved in hot hydrochloric acid, and titrated with permanganate of potassium.
[64] When chromium is present some of the iron may escape precipitation but it can be recovered from the solution by means of ammonic sulphide.
(1) 10FeSO4 + 2KMnO4 + 8H2SO4 = 5Fe2(SO4)3 + 2MnSO4 + K2SO4 + 8H2O. (2) 6FeCl2 + K2Cr2O7 + 14HCl = 3Fe2Cl6 + Cr2Cl6 + 2KCl + 7H2O.
(1) Fe2Cl6 + SnCl2 = 2FeCl2 + SnCl4.
(2) Fe2Cl6 + SH2 = 2FeCl2 + 2HCl + S.
(3) Fe2Cl6 + Na2SO3 + H2O = 2FeCl2 + Na2SO4 + 2HCl.
(4) Fe2Cl6 + Zn = 2FeCl2 + ZnCl2.
[67] 20 grams of stannous chloride and 20 c.c. of dilute hydrochloric acid are diluted to one litre.
[68] The maximum reducing effect of zinc is obtained by exposing as large a surface as possible of the metal in a hot concentrated solution containing but little free acid (Thorpe).
[69] About 5 inches in diameter.
[70] 61: 60:: 59: 58.13.
The iron in the ore is, then, the same in amount as that in 58.13 c.c. of the ferric chloride solution; and since 100 c.c. of the latter contain 1 gram of iron, 58.13 c.c. of the same contains 0.5813 gram of iron; and, further, if 1 gram of ore carries this amount of iron, 100 grams of ore will obviously give 58.13 grams of iron.
[71] These compounds are Ni2As and Co2As.
[72] With large quantities of iron the ferric precipitate should be re-dissolved and re-precipitated. The filtrate must be added to the original filtrate.
[73]
4KCy + NiSO4 = K2NiCy4 + K2SO4
2KCy + AgNO3 = KAgCy2 + KNO3
∴ 2AgNO3 = Ni
[74] Zn + H2SO4 = H2 + ZnSO4.
[75] These 104 c.c. are equivalent to 100 c.c. of dry air at 0° C. and 760 mm.
Tin occurs in nature as cassiterite (containing from 90 to 95 per cent. of oxide of tin), which mineral is the source from which the whole of the tin of commerce is derived. Tin also occurs as sulphide combined with sulphides of copper and iron in the mineral stannine or bell-metal ore. It is a constituent of certain rare minerals, such as tantalite.
The methods of assaying tin in actual use are remarkable when compared with those of other metals. The more strictly chemical methods are rendered troublesome by the oxide being insoluble in acids, resembling in this respect the gangue with which it is associated. Moreover, it is not readily decomposed by fusion with alkalies. The oxide has first to be reduced to metal before the tin can be dissolved. The reduction may be performed by fusing with potassic cyanide, by heating to moderate redness in a current of hydrogen or coal gas, or by heating to a higher temperature with carbon. The reduced metal is only slowly dissolved by hydrochloric acid, and although it is readily soluble in aqua regia, the solution cannot be evaporated or freed from the excess of acids, by boiling, without loss of tin, because of the volatility of stannic chloride. There has long been a difficulty in getting a quick wet method.
The process of assaying tin ores adopted in the mines of Cornwall is a mechanical one known as "vanning," the object of which is to find the percentage of "black tin," which, it is well to remember, is not pure cassiterite, much less pure oxide of tin. Tin ore, as taken from the lode, contains from 2 to 5 per cent. of cassiterite, and is mainly made up of quartz, felspar, chlorite, schorl, and other stony minerals, together with more or less mispickel, iron and copper pyrites, oxide of iron, and wolfram. The cassiterite has a specific gravity (6.4 to 7.1) considerably higher than that of the vein-stuff (2.5 to 3.0), and is concentrated by a series of washings till it is free from the lighter material. Those minerals which have a specific gravity approaching that of[Pg 272] the cassiterite are not completely removed. The mispickel and copper and iron pyrites are converted into oxides by roasting, and are in great part removed by a subsequent washing. The concentrated product is known as "black tin," and in this condition is sold to the smelter. The chief foreign matters in the black tin are silica, oxides of iron and copper, and wolfram, with traces of manganese and niobic acid; and in certain stream ores there may be as much as 6 or 7 per cent. of titaniferous iron. The black tin from the mines contains from 5 to 12 per cent. of water, and is sold and assayed wet. A series of typical samples of black tin ranged as follows:—
| Source of Material. | Percentage of Metal in Dry Ore. | Specific Gravity. |
| Good mine ore | 72.0 | 6.39 |
| Inferior do. | 71.5 | 6.64 |
| Titaniferous stream ore | 67.0 | 6.39 |
| Mine ore with wolfram | 64.5 | 6.67 |
| Ore from stream works | 58.5 | 5.99 |
It will be seen from these figures that black tin is a very variable substance; and that the specific gravity is largely influenced by the impurities; hence, it is only an indication of the percentage of metal when the same kind of ore is dealt with.
As already pointed out, the object of vanning is to determine the proportion of black tin in the lode stuff. The relation between the actual content in oxide of tin and the produce got by vanning has been tested on several occasions with results which show a fair degree of approximation.
The following are some published results of assays of the same batch of ore. The vanning results were obtained by a Cornish vanner of recognised ability, and the wet assays by two London firms of the highest standing:—
| Vanning results: | ||
| (Average) | 91 lbs. | of "black tin." |
| Wet Assay results: | ||
| A | 83.7 lbs. | of stannic oxide. |
| B | 79.7 lbs. | " |
The vanner reported his black tin as containing 70 per cent. of tin. This will bring his result, if calculated as stannic oxide, to 80.9 lbs. to the ton; which agrees with the others.
According to our experience the "van" assay agrees fairly well with the "wet" one, if the black tin is assumed to contain 92.5 per cent. of stannic oxide (SnO2).
Vanners are, as a rule, skilful men, and show remarkable dexterity in separating the black tin, with the help of their[Pg 273] apparatus, which consists simply of a shovel and a kieve of water. An account of the process is given below. But different vanners, all good men, will get different results working on material new to them. The black tin weighed by the vanner is supposed to correspond in quality with the black tin returned from the floors of the mine for which he is assaying, but this differs materially in different mines with the nature of the gangue. The process leaves too much to the judgment of the vanner. It is more than probable that in practice the returns from the dressing-floors check the assayer, instead of, as should properly be the case, the assayer checking the returns. It is only when this last is done that any control is had over the system of dressing. A correct assay of this ore is a matter of some importance, because of the high price of the metal.
The method of assaying the black tin is a dry one, and consists of mixing it with "culm," and submitting it in a black-lead crucible to the highest temperature of a wind furnace. The sample is taken wet as it arrives at the smelting house, and is assayed direct. The product of the assay is examined, and a deduction of a considerable percentage is very properly made for impurities, since the assay really determines the percentage, not merely of tin, but of the bodies present which are reducible at a white heat. The judgment as to how much is to be deducted is assisted partly by an examination of the metal got from the assay, and partly by the experience acquired in smelting similar ores. The produce, which is that of the impure tin, is stated in parts in twenty; thus a produce of 14 is equivalent to 70 per cent., or to 14 cwt. per ton.
This process, which has already been referred to, is carried out as follows:—After sampling the ore in the ordinary way, a quantity (varying with its richness) is weighed out. Special weights are generally used. The standard weight, marked 200, weighs about an ounce; with poor ores this quantity is taken for an assay, but with richer ores 100 or even 50 is sufficient. The unit of weight has no special name, but the parts in 200 are spoken of as the produce; thus, if 200 of ore were taken and 9.5 of black tin were separated, the produce would be 9-1/2: obviously half the "produce" will give the percentage. The weighed portion of the ore is placed on the vanning shovel. The vanner stands in front of a tub of water (kieve) and allows 30 or 40 c.c. of water to flow on to the ore. He then raises the shovel a little above the surface of the water, and, holding it nearly horizontal, briskly[Pg 274] rotates the water by imparting to the shovel a slight circular motion, passing into an elliptical one (front to back). This causes the finer mud to be suspended in the liquid, which is then run off, leaving the body of the ore in the centre of the shovel. This is repeated until the water after standing a moment is fairly clear. About half as much water as before is brought on; then, with a motion which is similar to the previous one, but with a jerk added in one direction, the heavier minerals are thrown up, and the stony matter brought back. The jerk is produced just as the wave of water is returning. The descending wave of water draws with it the bulkier and lighter particles of the ore, whilst the heavier matter lying on the bottom is scarcely affected by it. The jerky motion, however, carries it to the front of the shovel. The lighter stuff is washed off, and the residue dried by holding the shovel over the furnace. It now corresponds, more or less, to the stuff which on the mine is sent to the calciner. It is swept from the shovel into a scoop, and transferred to a hot crucible; in which it is calcined until free from sulphur. Some vanners calcine their samples before commencing to van. The calcined ore is shaken out of the crucible on to the shovel; rubbed up with a hammer; and washed (as at first) to get rid of the finer and lighter "waste." The separating motions are again gone through; and the "head" of the best of the black tin is thrown well up on one side of the shovel in the form of a crescent, so as to leave room on the shovel to work with the "tailings." The quantity of water used is kept low, to prevent this "crop" tin from being washed back again. The tailings are then crushed to free the tin from adherent oxide of iron; and again washed to throw up the remaining tin ore. As this tin is finely divided, it is more difficult to bring it up, so that a vigorous and rapid motion is required. The tailings are now washed off, and the whole of the black tin is brought into the centre of the shovel. It requires two or three washings more to free it from the waste it contains. Very small quantities of water are used. The purity of the black tin can be seen by its appearance on the shovel. The cleaned ore is dried as before, freed from particles of iron with the aid of a magnet, and weighed. The weighings are carried to 1/8th of the[Pg 275] unit used. The following example illustrates the method of calculation adopted on the mine. A parcel of 1 ton 2 cwt. 3 qrs. of tin ore with a produce of 45 (equal to 22-1/2 per cent.) contains 5 cwt. 0 qrs. 12 lbs. of black tin. This result is obtained as follows:—
ton cwt. qrs.
1 2 3
9 }
----------------- }
10 4 3 } equivalent to multiplying by 45.
5 }
---------------- }
5.1 3 3 strike off the first figure to the right.
4 multiply by 4 to reduce to quarters.
---------
4 12
3
---------
4 15
28 multiply by 28 to reduce to pounds.
-----
112
15
-----
12.7 strike off the first figure to the right.
Similarly, a parcel of 20 tons 10 cwt. with a produce of 9-1/2 contains 19 cwt. 1 qr. 25 lbs. of black tin. For the following information, as well as for much of that already given about vanning, we are indebted to Captain Reynolds, of Cook's Kitchen Mine. "To have a complete set of tools for all vanning purposes, it will be necessary to get the following:—A vanning shovel 14 inches long and 13 inches wide, weighing not over 2-3/4 pounds. It is made of hammered sheet iron of the shape shown in fig. 57. It must have a light wooden handle (preferably of deal) 3 feet long. A bruising hammer, weighing 2-1/2 pounds, with a handle 1 foot long. A pair of tongs (furnace) 2-1/2 feet long, made of 1/2-inch round iron. And a set of ordinary clay crucibles for calcining. There ought to be two sets of scales and weights: the first should be confined to weighing the powdered tin stuff, and the second ought to be a much higher class one, for weighing the black tin obtained. The furnace for roasting the sample should be 10 inches square and 12 inches deep, with the fire-bars at the bottom three-quarters of an inch apart. The water-box for vanning in should be at least 4 feet long, 2 feet 6 inches wide, and 8 inches deep."[Pg 276]
For the following description of the process adopted in Cornwall we are indebted to Mr. A.K. Barnett, F.G.S., of Chyandour.
Cornish Method.—Tin Ore Assay.—The ore to be smelted or assayed should be concentrated to say not less than 50 per cent. of metallic tin; though to obtain satisfactory results it should be brought nearer 70 per cent., as with ore containing less than 40 to 50 per cent. of metal there will be a considerable loss both in the assaying and in the smelting. If the ore to be operated on does not contain this quantity of metal, then the sample (if coarse) must be reduced to a fine state, the gangue being removed by vanning, and the ore saved for the fire assay.
The method adopted for the determination of tin in the ore is as follows:—About 2-1/2 ounces troy (1200 grains, or about 80 grams) of the ore to be assayed is weighed out and mixed on a flat copper pan (shaped with a long lip) with one-fifth of its weight (240 grains, or 15.5 grams) of powdered culm (anthracite). The mixture of ore and culm is either transferred to a black-lead crucible before the latter is put into the furnace, or, as some prefer, it is carefully swept into a crucible which has been imbedded in the fire. Some assayers cover their pots with a flat cover placed loosely on, while others leave the mixture in the open pot. The furnace, which has been previously fired to a strong heat, is then covered, and the sample is subjected to a sharp fire for a period of from twelve to twenty minutes. No definite time can be stated, as, besides the strength of the fire, the quality and condition of the ore, and the impurities associated with it, greatly affects the time required for the complete reduction of the ore. As soon as the mixture in the crucible has settled down to a uniform white heat, and any very slight ebullition which may have taken place has subsided, the crucible is gently shaken, removed from the fire (the culm-ash or slag which covers the metal being carefully drawn aside with an iron scraper), and the metal is poured quickly into an iron ingot-mould, which is usually placed on a copper pan to save the culm-slag and the adherent metal which comes out with it. The crucible is then carefully scraped, and the scrapings, together with the contents of the mould and pan, are transferred to a mortar. There the ingot of tin is freed from slag and then taken to the scales. The rest, after being finely powdered, is passed through a sieve. The flattened particles of tin which remain on the sieve are weighed with the ingot (the lump, as it is called); whilst the siftings are vanned on a shovel, and (the slag being washed off) the fine tin is collected, dried, and weighed with the rest: the whole gives the[Pg 277] produce or percentage of metal in the ore. The results of the assays are expressed in cwts. of metal in the ton of ore. The percentage is rarely given and never used in Cornwall. Thus—"13-1/2 Produce" would mean that the assay yielded results at the rate of 13-1/2 cwts. of metal for one ton of the ore. Some assayers use a little powdered fluor-spar to assist the fusion of refractory slags. A small quantity of borax will also occasionally be of service for ores containing silica in excess of any iron that may be present. The borax renders the slag more fusible, and assists the formation of a larger lump (with less fine tin in the slag) than would be obtained by the use of culm alone.
The quality and the percentage of pure tin in the metal will vary considerably, according to the impurities that are associated with the ore to be assayed.
The crude lump is then remelted in a small iron ladle at as low a temperature as possible, and the fused metal is poured into a shallow trench about 4 inches long by 3/4 of an inch wide cut in a block of white marble. The metal will be silvery-white if the temperature employed be correct; if too hot, the surface will show a yellow, red, or blue colour (according to the heat employed); in such case the metal should be remelted at a lower temperature. If the metal on cooling remains perfectly clear and bright, then it may be assumed that the tin is of good quality and commercially pure. A crystallised or frosted appearance of the metal indicates the presence of some alloy, say of iron, copper, zinc, lead, antimony, &c. The assayer who has had much practice can readily distinguish the metal or metals that are associated with the ore by noting the appearance of the tin on cooling; and can fairly judge the quantity of impurity present by the amount of the crystallisation or stain.
Whilst the foregoing method of assaying cannot lay claim to scientific accuracy, it is by no means so imperfect as some writers would have us believe, who state that a loss of 5 to 10 per cent. arises in the operation. It is certainly the most ready and expeditious mode of determining the commercial value of a parcel of tin ore, which, after all, is the main object of all assaying operations.
The difficulty which beginners find in obtaining satisfactory results, and any loss of metal which those not accustomed to the process may incur, will invariably occur in the vanning of the powdered slag for the fine tin, the rest of the operations being easy of execution, and requiring only the ordinary care necessary for all metallurgical work.
There is no doubt that if low percentage ores containing silica are assayed in this manner, low results are obtained, as it is impossible[Pg 278] to reduce the whole of the tin in the presence of free silica; with this class of ores, care should be taken to remove some of the silica by preliminary vanning, or some flux should be added which will combine with the silica, and so prevent its entering into combination with the tin. Low quality tin ores containing iron, copper, lead, zinc, antimony, etc., combined with arsenic, sulphur, or oxygen, will give very much higher results than the actual percentage of tin in the sample. The other metals (being readily reduced in the presence of tin) alloy with it, and give a hard lump difficult to fuse in the iron ladle; where the quantity of foreign metals is large, the metal can only be melted to a stiff pasty mass; so that (in determining the value of a ton of tin ore, or even reporting on the percentage of tin it contains) not only must the weight of the assay be the basis for calculation, but the quality and character of the metal obtained must also be considered. Thus two ores of tin might be assayed both yielding a similar produce, say 13-1/2 (67-1/2 per cent.), and yet one might contain 5 per cent. less tin than the other.
If it be required to obtain the pure metal from tin ores containing the ores of other metals associated with them, the latter must be removed by digesting in strong hydrochloric acid, and washing. The assay may then be conducted in the usual way, and a fairly pure lump will be obtained.
If wolfram be present in any appreciable quantity in the ore, it considerably reduces the proportion of lump, and at the same time it increases the fine tin (or prillion, as it is termed) in the assay. This may be got rid of by boiling in aqua regia, and dissolving out the tungstic acid which has been liberated by means of ammonia.
It will be seen that this method of assaying tin has its advantages and its drawbacks. It is quickly performed; with ores of good quality it gives results not to be excelled by any other process; and it gives the smelter the actual alloy and quality of metal he may expect to get in the smelting of the ore, which no other mode of assaying will do: against which may be set the skill required to obtain accurate results with the vanning shovel; the loss of metal in poor ores containing an excess of silica; and the high results from ores containing a large quantity of metallic impurities.
Cyanide Method.—Weigh up 20 grams of the ore and dry it on a scoop over the Bunsen flame. When dry, weigh, and calculate the percentage of water from the loss in weight. Transfer the dried ore to an evaporating dish, and cover with 30 c.c. of hydrochloric acid; boil for 10 or 12 minutes, and then add 5 c.c.[Pg 279] of nitric acid and boil again. Dilute with water, and filter. Transfer the filter and its contents to an E Battersea crucible, and calcine it for a few minutes. Cool, and weigh the residue. The loss equals the oxides soluble in acid. Transfer the residue to the crucible and mix it with its own weight of cyanide of potassium; add a similar amount of "cyanide" as a cover. Place in the furnace, and when the charge has attained the temperature of the furnace (in from 3 to 6 minutes), remove it at once; tap the pot vigorously several times, and then pour its contents quietly into a mould. Dissolve the slag in water, clean, dry, and weigh the button of tin.
Detection.—Tin ore is detected by its insolubility in acids, high specific gravity, and characteristic appearance in water. The powder is separated from the lighter gangue by washing. It is fused in a Berlin crucible with five times its weight of potassic cyanide at a moderately high temperature in a muffle, or over the blowpipe. The slag is washed off with water, and the metallic buttons or residue treated with hydrochloric acid (not aqua regia), for some time. One portion of the solution strikes a purple colour with chloride of gold, another portion gives a white or grey precipitate or cloudiness with mercuric chloride. These reactions are characteristic of tin as stannous chloride.
Metallic tin treated with nitric acid becomes converted into a white insoluble powder (metastannic acid). Aqua regia dissolves tin readily, forming stannic chloride, and in this solution the metal is detected by precipitation with sulphuretted hydrogen, which gives a yellow precipitate. Tin in solution as stannic or stannous chloride is precipitated as metal by means of zinc.
The fact that tin forms two well-defined series of compounds is taken advantage of in assaying (just as in the case of iron), by determining how much of an oxidising agent is required to convert it from the stannous into the stannic state. For example, on the addition of a solution of permanganate of potash to a solution of stannous chloride the oxidation goes on rapidly, and the finishing point is sharp and distinct; but acid solutions of stannous chloride quickly take up oxygen from that dissolved in the water used and from the air. Unfortunately, there is no obvious sign that such oxidation has taken place, except that (fatal to the assay) a smaller volume of the permanganate is required. Great care is required with such solutions, both before and during titration. The addition of an excess of ferric chloride to the stannous[Pg 280] solution, as soon as the whole of the tin has been dissolved, will lessen this liability to oxidation.
Separation.—If the tin is present in an alloy, the substance is boiled in an evaporating dish with dilute nitric acid until the whole of the material is attacked. Evaporate nearly to dryness, dilute, boil for a few minutes, and filter off the white insoluble residue. Under certain circumstances this residue will be nearly free from other metals, in which case it is ignited and weighed. If not known to be pure it must be ignited, reduced in a current of hydrogen, and treated as subsequently described.
When the tin is present as insoluble oxide in an ore, the substance is finely powdered, and from 1 to 5 grams of it (according to its richness) boiled with 30 c.c. of hydrochloric acid in an evaporating dish till the oxide of iron is seen to be dissolved. Then add 1 c.c. of nitric acid (or more if much pyrites, &c., is present) and continue the boiling till these are decomposed; dilute and filter off, washing first with dilute acid and afterwards with a little dilute ammonia, dry, ignite, and place in a combustion tube (together with the filter-ash) and heat to redness for about thirty minutes in a current of dried hydrogen.

The oxide of tin is placed in a porcelain boat (fig. 58), which is then introduced into a piece of combustion tube. The latter, wrapped in a piece of wire gauze, is supported on a couple of iron rings, and heated by one or two Bunsen burners in a furnace fitted up with loose fire-brick tiles, as shown in fig. 59.
When the reduction is complete the tube is allowed to cool; the boat is removed and the tin dissolved. Add a rod of zinc to the freely-acid hot solution, and in a few minutes decant through a filter and wash with water, after having removed the zinc. Wash the precipitated metal back into the beaker, and dissolve in 10 c.c. of dilute nitric acid, evaporate off the excess of acid;[Pg 281] dilute, boil, and filter. Wash, dry, ignite strongly in a porcelain crucible, and weigh.
In the absence of antimony the above separation works very well, but if this metal is present in quantity the metals precipitated on the zinc must be covered with hydrochloric acid and treated with a few drops of nitric. It is then warmed with iron wire until no more of the latter dissolves. The antimony is precipitated as metal, and the tin remains in solution as stannous chloride. The antimony is filtered off, and may be washed with alcohol, and weighed, whilst the tin in the filtrate is precipitated with zinc, and treated as already described.
If the tin is not already in the metallic state it is reduced to this condition by the method given (precipitation by zinc). Treat the finely-divided metal (washed free from chlorides) in a four-inch evaporating dish with 10 c.c. of dilute nitric acid, cover with a clock-glass, and apply a gentle heat until the precipitate appears of a white colour and the metal is completely attacked. Evaporate nearly to dryness on a water-bath; then add 50 c.c. of water, heat to boiling, and filter. Wash with hot water, dry, transfer to a weighed porcelain crucible, add the filter-ash, ignite strongly, and weigh. The precipitate after ignition is stannic oxide (SnO2). It is a yellowish-white powder (darker whilst hot), insoluble in acids, and contains 78.67 per cent. of tin. Cold dilute nitric acid dissolves tin to a clear solution, which becomes a white enamel-like jelly on heating; this (filtered off, washed, and dried) forms an opal-like substance, which is converted on ignition into stannic oxide with evolution of nitrous fumes. Stannic oxide when ignited with chlorides is more or less completely converted into stannic chloride, which volatilises. The presence of chlorides during the evaporation with nitric acid causes a similar loss.
Determination of Tin in an Alloy.—(Bronze.)—Take 2 grams, and attack with 20 c.c. of dilute nitric acid in a covered beaker with the aid of heat. Boil till the bulk is reduced by one-half, dilute with 50 c.c. of water, allow to settle for a few minutes, and filter; wash well first with water acidulated with a little nitric acid, and afterwards with water; dry, ignite, and weigh as stannic oxide.
Determination of Tin in Tin Ore.—Treat 5 grams of the dried and finely-powdered ore with 30 c.c. of hydrochloric acid in a four-inch evaporating dish. After the soluble oxides have been dissolved add 1 or 2 c.c. of nitric acid, boil off nitrous fumes,[Pg 282] dilute, and filter. Dry the filter, transfer the cleaned ore to a piece of combustion tube ten or twelve inches long and narrowed at one end. Pass a current of hydrogen through the tube and heat to redness for 30 minutes; cool whilst the gas is still passing. Dissolve in 20 c.c. of dilute hydrochloric acid and keep the solution tinted with permanganate of potassium. When the colour of the permanganate becomes permanent dilute to a bulk of 50 c.c. with water, filter, and wash. Heat; add a rod of zinc weighing about 3 grams; allow to stand for a few minutes; decant through a filter; and wash, removing the remaining zinc and returning the tin to the beaker. Treat with 5 c.c. of dilute nitric acid, boil for some time, take up with water, filter, wash, dry, ignite, and weigh as stannic oxide.
Titration with Solution of Permanganate of Potassium.—This titration may be made either directly on the solution of stannous chloride (prepared by dissolving the precipitated metal in hydrochloric acid), or indirectly, on a solution of ferrous chloride (produced by the reducing action of the precipitated metal on ferric chloride). The standard solution of permanganate of potassium is made by dissolving 5.356 grams of the salt in water and diluting to one litre. 100 c.c. are equivalent to 1.00 gram of tin.
The precipitated tin is transferred to a flask; and dissolved in 10 c.c. hydrochloric acid, with the aid of heat and in an atmosphere of carbonic acid. The acid and metal are placed in the flask; which is then filled with the gas, and stopped with a cork provided with a rubber valve. When solution is complete the flask is again filled with carbonic acid. Fifty c.c. of water freed from air and saturated with carbonic acid are then added. This water is made by adding a gram of bicarbonate of soda and 2 c.c. of hydrochloric acid to 100 c.c. of water: the effervescence sweeps out the dissolved oxygen. The permanganate of potassium solution is then run in from a stop-cock burette in the usual way until a faint pink tinge is obtained.
The following experiments show the effect of variations in the conditions of the assay. A solution of stannous chloride equivalent in strength to the "permanganate" was made by dissolving 19.06 grams of the crystallised salt (SnCl2.2H2O.) in 50 c.c. of water and 10 c.c. of hydrochloric acid and diluting to 1 litre with water freed from dissolved oxygen. 100 c.c. contain 1 gram of tin. In the first experiments tap water was used and no precautions were taken for excluding air. Except when otherwise[Pg 283] stated, 20 c.c. of the stannous chloride were used in each experiment with 10 c.c. of hydrochloric acid, and were diluted to 100 c.c. with water before titration.
Effect of Varying Hydrochloric Acid.
| Acid added | 1.0 | c.c. | 10.0 | c.c. | 20.0 | c.c. | 50.0 | c.c. |
| "Permanganate" required | 18.8 | " | 18.9 | " | 18.8 | " | 18.8 | " |
The only effect of the increase in quantity of acid was to give the brown of perchloride of manganese instead of the pink of permanganic acid to mark the finishing point.
Effect of Varying Temperature.
| Temperature | 15° C. | 50° C. | 70° C. | 100° C. |
| "Permanganate" required | 18.8 c.c. | 18.7 c.c. | 18.6 c.c. | 18.4 c.c. |
Rate of Atmospheric Oxidation.—Solutions ready for titration were exposed to air at the ordinary temperature for varying lengths of time and then titrated.
| Time exposed | 0 min. | 5 min. | 10 min. | 20 min. | 60 min. |
| "Permanganate" required | 18.8 c.c. | 18.8 c.c. | 18.8 c.c. | 18.8 c.c. | 18.6 c.c. |
It is best to titrate at once, although the loss by oxidation is only small after one hour's exposure.
Effect of Varying Tin.
| Stannous chloride added | 1.0 | c.c. | 10.0 | c.c. | 20.0 | c.c. | 50.0 | c.c. | 100.0 | c.c. |
| "Permanganate" required | 0.7 | " | 8.8 | " | 18.0 | " | 47.4 | " | 95.4 | " |
Effect of Varying Bulk.
| Bulk | 50.0 | c.c. | 100.0 | c.c. | 200.0 | c.c. | 500.0 | c.c. |
| "Permanganate" required. | 9.0 | " | 18.3 | " | 17.4 | " | 15.1 | " |
The two last series show an interference, which is due to the oxygen dissolved in the water, as may be seen from the following similar experiments, which were, however, performed with water freed from oxygen and in which the titrations were effected in an atmosphere of carbonic acid.
Effect of Varying Tin.—A new solution of stannous chloride was used.
| Stannous chloride added | 1.0 | c.c. | 10.0 | c.c. | 20.0 | c.c. | 50.0 | c.c. | 100.0 | c.c. |
| "Permanganate" required | 1.0 | " | 10.0 | " | 19.8 | " | 49.6 | " | 99.3 | " |
Effect of Varying Bulk.
| Bulk | 30.0 | c.c. | 50.0 | c.c. | 100.0 | c.c. | 200.0 | c.c. | 500.0 | c.c. |
| "Permanganate" required | 19.8 | " | 19.8 | " | 19.8 | " | 19.8 | " | 19.8 | " |
It will be seen that in working under these conditions the results are proportional and the method satisfactory.
Examination of Tin Phosphide.—(Phosphor Tin.)—This substance is used in the manufacture of "phosphor bronze" and similar alloys. It is a crystalline, imperfectly-malleable, metallic substance. It is soluble in hydrochloric acid with effervescence; phosphoretted hydrogen, which inflames on the addition of a drop or two of nitric acid, being evolved. It is attacked by nitric acid, yielding a white powder of stannic phosphate; this is not easily decomposed by ammonium sulphide or readily soluble in hydrochloric acid.
"Phosphor-tin" is made up only of tin and phosphorus. For the estimation weigh up 1 gram. Place in a weighed Berlin dish; and cover with 10 c.c. of nitric acid and 3 or 4 c.c. of water. Let the reaction proceed (under a clock-glass) on the water-bath till complete. Remove the glass; evaporate to dryness, and ignite, at first gently over a Bunsen burner, and afterwards in the muffle at a red heat. Cool in the desiccator, and weigh as quickly as possible when cold. The substance contains the tin as stannic oxide, SnO2, and the phosphorus as phosphoric oxide, P2O5. The increase in weight on the gram of substance taken gives the weight of the oxygen taken up by the phosphorus and tin, and since 1 gram of tin takes up only 0.271 gram of oxygen, and 1 gram of phosphorus takes up 1.29 gram, the proportion of tin to phosphorus can be calculated from the increase in weight. For example, 1 gram of a sample gave 1.3410 gram of mixed oxides, which is 0.070 gram in excess of that which would be got with pure tin. If the substance was all phosphorus the excess would be 1.0190 gram; consequently the proportion of phosphorus in the substance is 0.070 / 1.019, or 6.87 per cent. The tin is calculated by difference, 93.13 per cent.
Another method of separating and determining the phosphorus is as follows:—Take 1 gram of the substance and add to it 15 c.c. of hot aqua regia. Boil till dissolved, dilute, and precipitate the tin with sulphuretted hydrogen. To the filtrate add ammonia and "magnesia mixture." Filter; wash the precipitate with dilute ammonia; dry, ignite, and weigh as magnesic pyrophosphate. Calculate the phosphorus, and take the tin by difference.
A sample of phosphor tin gave—
| Tin | 93.1 | per cent. (by difference) |
| Phosphorus | 6.9 | " |
| ——- | ||
| 100.0 |
Tin Arsenide.—This is met with in tin-smelting; it closely resembles the phosphide, but the crystals have a duller grey[Pg 285] appearance. It contains simply tin and arsenic. The determination is made by treating 1 gram of the substance with nitric acid and weighing the mixed oxides of tin and arsenic in the same manner as in the case of the phosphide. One gram of arsenic will give 1.533 gram of arsenic oxide, As2O5; consequently the excess of weight of the mixed oxides over 1.271 gram must be divided by 0.262; the result multiplied by 100 gives the percentage of arsenic. In consequence of the higher atomic weight of arsenic the results by this method are not so close as with the phosphide. Each milligram of excess weight (over 1.271) represents 0.38 per cent. of arsenic, As. Both in this and in the corresponding phosphide determination care must be taken to avoid absorption of moisture, by allowing the oxides to cool in a desiccator and weighing quickly.
The percentage of arsenic is better determined as follows:—Weigh up 1 gram of the substance, dissolve in aqua regia, dilute, and pass sulphuretted hydrogen. Render alkaline with ammonia, and add ammonium sulphide till the precipitate is dissolved. Add "magnesia mixture." Filter off the precipitate, wash with dilute ammonia, ignite with a few drops of nitric acid, and weigh as magnesic pyrarsenate. Calculate the arsenic and take the tin by difference. A sample treated in this way gave—
| Tin | 96.8 | per cent. by difference |
| Arsenic | 3.2 | " |
| ——- | ||
| 100.0 |
Examination of Black Tin.—Dry the ore, and reduce it to a fine powder. Weigh up 2 grams, and boil with 20 c.c. of hydrochloric acid and 2 c.c. of nitric for ten or fifteen minutes. Filter, and reserve the filtrate.
Tungstic Acid.—Digest the residue with about 50 c.c. of water and a few c.c. of dilute ammonia for a few minutes, and filter; collect the filtrate in a weighed porcelain dish, evaporate to dryness, ignite, and weigh as tungstic acid, WO3.
Stannic Oxide.—Dry, ignite, and weigh the insoluble residue. Transfer to a porcelain boat, and reduce in a current of hydrogen at a red heat for half an hour. Allow to cool whilst the hydrogen is still passing. Transfer the boat to a beaker, and dissolve up the tin in 10 c.c. of hydrochloric acid and a c.c. or so of nitric. Wash out the combustion tube with some acid and add the washing to the contents of the beaker. Warm gently, dilute with water, and filter. Collect, dry, ignite, and weigh the insoluble residue. Through the filtrate pass a rapid current of sulphuretted hydrogen, allow to settle, and filter. Wash the precipitate with hot water, dry, calcine gently; ignite with ammonium [Pg 286]carbonate, and weigh as stannic oxide, SnO2. The insoluble residue will in most cases retain some tin. Fuse it with fusion mixture, take up with hydrochloric acid, filter, pass sulphuretted hydrogen through the filtrate, collect and wash the sulphide of tin. Ignite and weigh as stannic oxide, and add it to that previously obtained.
Copper.—Pass sulphuretted hydrogen through the acid filtrate obtained in the first cleaning of the ore, collect the precipitate, and wash first with soda solution and then with hot water. Dry, ignite, and weigh as cupric oxide, CuO. Mix the filtrate with that from the main portion of the sulphide of tin.
Ferric Oxide.—Boil off the sulphuretted hydrogen from the mixed filtrates and peroxidise with nitric acid. Add ammonia in slight excess, boil, filter, dry, ignite, and weigh the precipitate as ferric oxide. This will be practically pure, but the iron in it must be determined by dissolving and titrating. The filtrate from the iron may contain zinc, lime, and magnesia, but rarely in quantities sufficient to be determined.
Silica, &c.—-The silica may be calculated from the weight of the residue insoluble in acid, after the reduction of the tin in hydrogen, by deducting from it the weight of the oxide of tin subsequently found. Or it may be determined as follows:—The insoluble portion is fused with fusion mixture, and taken up with hydrochloric acid, as already described. On filtering, the filter will retain a portion of the silica. The rest is recovered, after the removal of the stannous sulphide, by evaporating to dryness, taking up with hydrochloric acid, and filtering through the same filter. It is washed, dried, ignited, and weighed as silica. The filtrate from the silica is boiled with a little nitric acid and precipitated with ammonia. The precipitate is collected, washed, ignited, and weighed as ferric oxide and alumina (but it frequently contains oxide of titanium). When the last is present it is determined by fusing with bisulphate of potash and extracting with cold water. The solution is nearly neutralised with ammonia, charged with sulphurous acid, and boiled. The precipitate is collected, washed, dried, ignited, and weighed as oxide of titanium, TiO2. The difference between this weight and that of the combined oxides gives the ferric oxide and alumina. The filtrate from the mixed oxides is examined for lime and magnesia.
Sulphur.—Rub up 5 grams of the ore with 5 grams of nitre, transfer to a porcelain dish, and fuse over a Bunsen burner for fifteen minutes. When cold, extract with water, and determine the sulphur volumetrically with standard barium chloride. The sulphur may be present as sulphide or sulphate.
Arsenic.—Take 5 grams, and evaporate with nitric acid; dilute, add ammonia, pass sulphuretted hydrogen, and filter.[Pg 287]
To the filtrate add "magnesia mixture." Collect the precipitate, ignite with nitric acid, and weigh as magnesic pyrarsenate.
The following may be taken as an example of the composition of an impure black tin:—
| Tungstic acid | 1.8% |
| Stannic oxide | 79.0 |
| Silica | 2.6 |
| Titanic oxide | 0.8 |
| Copper oxide | 0.9 |
| Ferric oxide | 13.4 |
| Sulphur | 0.4 |
| Arsenic | 0.3 |
| —— | |
| 99.2 |
Examination of Hardhead.—In the smelting of tin ores a quantity of speise, known as "hardhead," is produced. It is essentially an arsenide of iron, carrying a considerable quantity of tin. Much of this last is present in the form of small buttons of metal distributed through the mass. The buttons can be seen on careful inspection, and become evident on powdering.
In assaying the substance, a variation in the usual method of sampling is required, because of the quantity of metal present which cannot be powdered. After powdering as finely as possible, the coarse particles are sifted off and weighed. The weight of the powder is also taken. The method of working is best illustrated by an example. A sample of hardhead weighed 155.1 grams, and gave 21.0 grams of coarse particles, equivalent to 13.5 per cent. of the whole. The fine portion weighed 134 grams, which is equivalent to 86.5 per cent.
Thirteen and a half grams of the coarse material were dissolved in aqua regia, and diluted with water to 1 litre. Ten c.c. of this contain 0.135 gram of the metallic portion, which is the amount contained in 1 gram of the original hardhead. If, in a determination, 1 gram of the substance is wanted, weigh up 0.865 gram of the powdered portion, and add to it 10 c.c. of the solution. It will be seen that these together make up 1 gram of the original sample. The solution of the metallic portion must be saved until the analysis is finished.
Tin and Copper.—Weigh up the portion of the powdered stuff equivalent to 1 gram of the sample. Transfer to a flask, and cover with 10 c.c. of the solution of the metallic portion and 10 c.c. of aqua regia. Boil gently till oxidation is complete and the nitric acid for the greater part driven off. Dilute to 100 c.c. with water, and pass sulphuretted hydrogen for some time. Filter, wash with hot water, and rinse through the funnel back into the flask. Digest with yellow sodium sulphide until only a[Pg 288] light, flocculent, black precipitate is left. Filter this off, wash with hot water, dry, calcine, treat with a little nitric acid, ignite, and weigh as copper oxide, CuO. The weight multiplied by 0.7983 gives the weight of copper.
The filtrate containing the tin is rendered acid with hydrochloric acid, and filtered. The precipitate is rinsed into a half-pint beaker, covered with 20 c.c. of hydrochloric acid, and boiled down to about 20 c.c. The solution is filtered off from the sulphur and sulphide of arsenic, which, after washing with hot water, is transferred to a flask labelled "arsenic." A strip of sheet zinc (2 in. by 1 in.) is placed in the solution. The evolution of hydrogen should be brisk. In five or ten minutes decant off a few c.c. of the liquid, and test with sulphuretted hydrogen for tin. If no yellowish precipitate is formed, decant off the rest of the liquid, and wash the precipitated metal with hot water two or three times by decantation. The metal should be in a lump; if there are any floating particles they must be made to sink by compression with a glass rod. Transfer the washed metal to an evaporating dish 3 or 4 in. across, and cover with a few c.c. of hot water. Add nitric acid drop by drop till the tin is completely attacked. Evaporate nearly to dryness, and add a drop or two more of nitric acid and 20 c.c. of water. Boil and filter. Wash with hot water, dry, ignite, and weigh as stannic oxide, SnO2. Calculate to metallic tin by multiplying by 0.7867.[76]
The filtrate from the first treatment with sulphuretted hydrogen will probably no longer smell of the gas. Warm and pass the gas for a few minutes longer. Filter off any precipitate of sulphide of arsenic, and transfer it to the flask for "arsenic." Boil the filtrate (ignoring any signs of a further precipitation of arsenic) with a few c.c. of nitric acid, and separate the iron as basic acetate. Wash; reserve the filtrate for cobalt.
Iron.—Rinse back the "basic acetate," precipitate into the flask, add ammonia, dilute with water to about 100 c.c., and pass sulphuretted hydrogen for a few minutes. Filter, and wash with hot water. Collect the filtrate in the flask labelled "arsenic." Boil the precipitate with dilute sulphuric acid, filter, and titrate the filtrate with the permanganate of potassium solution after boiling off the sulphuretted hydrogen. Report the result as iron. The sulphuric acid will not effect complete solution, a light black residue will remain, chiefly sulphur; this must be rinsed into the filtrate from the acetate separation. It contains cobalt.
Cobalt.—The filtrate from the acetate separation will have a pink colour. Render it ammoniacal and pass sulphuretted[Pg 289] hydrogen. Collect the precipitate on a filter, dry, and ignite. Dissolve in hydrochloric acid, and evaporate nearly to dryness with an excess of nitric acid. Dilute with 10 or 20 c.c. of water and add potash solution in slight excess. Add acetic acid until the solution is acid and the precipitate is quite dissolved. Add 20 or 30 c.c. of a strong solution of potassium nitrite, and determine the cobalt, as described on pp. 254, 256. Boil the filtrate from the cobalt, precipitate with hydrochloric acid, render ammoniacal, and test for zinc, nickel, and manganese.
The remainder of the tin will be contained in the flask labelled "arsenic." Acidify with hydrochloric acid and filter. Rinse into a beaker, and evaporate to a small bulk with 10 c.c. of nitric acid. Dilute and filter. Dry the precipitate, consisting of stannic arsenate (2SnO2.As2O5), ignite, and weigh. Calculate the tin it contains by multiplying by 0.4453, and add to that already found.
Arsenic.—This is determined in a separate portion. Weigh up a portion of the powder equivalent to 1 gram of the hardhead, place in a pint flask, and boil with 10 c.c. of nitric acid. When action has ceased add 10 c.c. of the solution of the metallic portion and then hydrochloric acid (a few drops at a time) till solution is complete. Warm gently in dissolving, but do not boil. Dilute to about 100 c.c., render alkaline with ammonia, and add 20 c.c. of yellow ammonium sulphide. Digest at a gentle heat for about thirty minutes, filter, and wash. Add 50 c.c. of magnesia mixture, shake well, allow to stand for an hour, filter, and wash with dilute ammonia. The precipitate is dissolved and then titrated with uranium acetate, or it is evaporated with nitric acid, ignited, and weighed as pyrarsenate of magnesia. Calculate the result to arsenic, As.
Sulphur.—Weigh up a portion of the powder equivalent to 2 or 3 grams of the hardhead. Rub up in a mortar with 5 grams of nitre and fuse in a porcelain dish for ten minutes. Extract with water, add 20 or 30 c.c. (as the case may be) of the solution of the "metallics." Add 10 grams of sodic acetate, and ferric chloride until the precipitate turns brown; dilute with water to half a litre, boil, and titrate with standard baric chloride, as described under Sulphur. Report as sulphur.
A sample of hardhead examined in this way gave—
| Sulphur | 3.00% |
| Arsenic | 27.10 |
| Tin | 22.2 |
| Copper | 1.64 |
| Iron | 43.2 |
| Cobalt | 2.6 |
| ——— | |
| 99.74 |
Examination of Tin Slags.—In tin smelting works the term "slag" is applied to the unfused portion of the charge. It is made up of unburnt anthracite and small lumps of slag proper together with some buttons of metallic tin. This is rarely, if ever, assayed. The slag proper (or, as it is generally called, "glass") is a silicate of iron, alumina, and lime, containing from 3 to 7 per cent. of tin. It is thus examined:—The sample after bruising on an iron plate, is reduced to a very fine powder by grinding in an agate mortar. In this state it is in most cases readily decomposed by hydrochloric acid.
Determination of Tin.—Where the percentage of tin only is required, take 2 grams of the powdered slag and well mix with it 20 c.c. of hydrochloric acid, and heat to boiling. Add 1 c.c. of nitric acid, allow to stand for fifteen minutes, dilute with water, and filter. Pass a rapid current of sulphuretted hydrogen for some time. Allow to settle, and filter. The precipitate, after washing with hot water, is dried, and gently calcined until the greater part of the sulphur is burnt off. It is then strongly ignited in the muffle (or over the blowpipe) with the addition of a small lump of ammonic carbonate. The residue is weighed as stannic oxide (SnO2); and is calculated to metallic tin by multiplying by 0.787. The percentage on the slag is calculated in the usual way.
The tin is always best determined in the examination of slags by a separate assay carried out in this way. The determination of the other constituents is thus made:—
Silica.—Take 2 grams of the powdered slag and cover them, in a small evaporating dish, with 20 c.c. of hydrochloric acid; mix well by stirring with a glass rod; and evaporate to dryness. If (as is generally the case) tungsten is present the solution will be blue. Take up with 20 c.c. of hydrochloric acid. Add 1 c.c. of nitric acid; and reduce by boiling to about half the bulk. Add about 20 c.c. of water, boil, and filter. Wash the residue with hot dilute hydrochloric acid. It consists of silica with the tungstic acid. Wash it back into the dish; and digest with 5 or 10 c.c. of a cold solution of ammonic carbonate. Filter; and collect the filtrate and washings in a weighed porcelain dish. Dry the residue, ignite strongly, and weigh as silica, SiO2. In certain exceptional cases this may contain some unaltered cassiterite, which is easily recognised by its appearance.
Tungsten.—The ammonic carbonate filtrate from the silica is evaporated to dryness, ignited strongly over the blowpipe, and weighed. The residue is tungstic acid, WO3. The tungsten may be conveniently reported in this form, although it is probably present as a lower oxide.[Pg 291]
Tin.—The acid filtrate from the silica and tungstic acid is treated with sulphuretted hydrogen. The sulphide of tin is filtered off. Since the percentage of tin has been already determined, this precipitate may be neglected; or may be treated in the same way as the previous one, so as to check the result. Since some stannic chloride will have been lost in the evaporation, a low result may be expected. The tin should be reported as stannous oxide; and is calculated by multiplying the percentage of tin by 1.136.
The filtrate from the tin is boiled rapidly down to remove sulphuretted hydrogen; and then peroxidised with 1 or 2 c.c. of nitric acid. It is cooled, transferred to a graduated flask, and diluted with water to 200 c.c.
Ferrous Oxide and Alumina.—Half the filtrate from the tin (that is, 100 c.c.) is taken, nearly neutralised with soda, and treated with sodium acetate. The basic acetate precipitate obtained on boiling is filtered off and washed. Reserve the filtrate. The precipitate is dissolved off the filter with hot dilute hydrochloric acid; and the solution thus formed is treated with a slight excess of ammonia, and boiled. The precipitate is filtered off, washed with hot water, dried, ignited, and weighed as mixed ferric oxide and alumina. The ignited precipitate is then dissolved with sulphuric and hydrochloric acids; and the iron determined in the solution by titration with the solution of stannous chloride. The iron found is calculated to and reported as ferrous oxide, FeO (factor = 1.286). To find the alumina, which is best estimated by difference, multiply the iron by 1.428 to get the weight of ferric oxide, and deduct this from the weight of alumina and ferric oxide found. This, of course, gives the alumina. A direct determination may be made by removing the tin from the titrated solution with sulphuretted hydrogen, filtering, nearly neutralising with ammonia, and boiling with a few grams of hyposulphite of soda. The precipitate, filtered, washed, and ignited, is the alumina, which is weighed. The direct determination gives a slightly low result.
Oxides of Zinc and Manganese.—These are determined in the filtrate from the basic acetate precipitate by rendering alkaline with ammonia, and passing a current of sulphuretted hydrogen. Generally a small, but decided, precipitate of alumina comes down, together with sulphides of any zinc or manganese which is present. The precipitate is allowed to settle, dried, ignited, and weighed. The metals are separately determined in it; and the residue is counted as alumina, and added to that already found. The mixed precipitate amounts to from 1 to 2 per cent. of the sample.[Pg 292]
Lime.—The filtrate from the last is treated with ammonic oxalate, boiled for a few minutes, allowed to settle, and filtered. The precipitate is washed with hot water; dried; ignited; and weighed as carbonate, after gentle ignition; or as lime, after strong ignition in the muffle.
Magnesia.—The filtrate from the lime is treated with sodic phosphate and ammonia. It is well mixed by stirring, and allowed to stand overnight. The precipitate is washed with dilute ammonia, dried, ignited, and weighed as pyrophosphate.
Soda and Potash.—These are determined in the remaining half of the filtrate from the tin. The solution is rendered ammoniacal with ammonia; and treated, first with sulphuretted hydrogen, and then with ammonium oxalate. The precipitate is filtered off and rejected. The filtrate is evaporated in a small porcelain dish over a Bunsen burner, or on the sand bath; and towards the close (or earlier if the evaporation is not proceeding well) nitric acid is added. The evaporation is carried to dryness; and the residue heated nearly to redness. The residue, which consists of magnesia with carbonates and chlorides of the alkalies, is extracted with water; and filtered. The filtrate is evaporated with hydrochloric acid in a weighed platinum dish, ignited gently, and weighed. This gives the weight of the mixed chlorides of sodium and potassium; which are then separated and determined as described under Potash.
It must be remembered when calculating the percentage that (with the exception of the silica, tungstic acid, and tin) the determinations have been made on 1 gram of the sample.
The following analysis will illustrate the composition of such a slag:—
| Tungstic acid | 1.3% |
| Silica | 39.4 |
| Stannous oxide | 8.1 |
| Ferrous oxide | 26.2 |
| Alumina | 14.8 |
| Oxide of manganese | traces |
| Lime | 7.9 |
| Magnesia | 0.5 |
| Alkalies calculated as soda | 1.7 |
| ——— | |
| 99.9 |
Titanium only occurs as a mineral in its oxidised state, or as titanic oxide (TiO2). It is a substance which has little commercial value, and is generally recognised as one of the rare bodies; although, in small quantities, it is widely disseminated. It occurs in granite, basalt, and other igneous rocks in quantities up to as[Pg 293] much as 1 per cent. It is also met with in clays and iron ores, and in river sands, in which it is often associated with stream tin. The proper minerals of titanium are rutile (TiO2), titaniferous iron (titanate of iron), and sphene (titanate and silicate of lime).
The oxide of titanium (like cassiterite and quartz) is undecomposed by hydrochloric or nitric acid; so that it is generally found in the residue insoluble in acids. The titanates, however, are attacked, and a portion of the titanium dissolves; so that it must be looked for in both the filtrate and residue. Oxide of titanium in its native form, or after ignition, may be made soluble by fusing the finely-divided substance with fusion mixture in a platinum dish. The resulting titanate is dissolved out of the "melt" by cold hydrochloric acid.
The method most commonly used is fusion with bisulphate of potash. This renders the oxide of titanium soluble in cold water. The process is as follows:—The substance is extracted with hydrochloric and nitric acids, and the solution reserved for further treatment; the residue is dried, moistened with sulphuric acid, and evaporated once or twice to dryness with hydrofluoric acid. It is then fused with bisulphate of potash, and the "melt" extracted with cold water until all soluble matter is removed. The solution is filtered. The residue may consist of unremoved silica, and oxides of tantalum, niobium, and, perhaps, chromium. On the prolonged boiling of the filtrate, the oxide of titanium (and oxide of zirconium, if any) is precipitated.
Any titanium dissolved by the first extraction with acids is recovered in the following way:—Sulphuretted hydrogen is passed into the acid solution, and any precipitate that may be formed is filtered off. The filtrate is oxidised, and the iron, aluminium, and titanium are separated as basic acetates (see under Iron). The precipitate is dried and fused with bisulphate of potash. The "melt" is extracted with cold water, filtered if necessary, and the solution rendered first faintly alkaline with ammonia, then very slightly acid with sulphuric acid. 30 or 40 c.c. of a saturated solution of sulphurous acid is added, and the oxide of titanium precipitated by prolonged boiling. It is filtered off, added to the precipitate previously got, ignited with ammonic carbonate towards the end, and then weighed.
Detection.—Titanium is detected in an insoluble residue by fusing the residue for some time in a bead of microcosmic salt. In the reducing flame it gives a violet colour, which becomes reddish-brown if much iron is present. In the oxidising flame it gives a colourless or whitish bead. It is best detected in acid solutions by the deep brown or iodine colour developed on adding hydroxyl. A solution of this can be prepared [Pg 294]by pouring peroxide of barium (BaO2) diffused in water into dilute hydrochloric acid (a little at a time), and keeping the acid in excess.
Separation.—In the usual course of an analytical separation the hydrate of titanium will be thrown down with ferric hydrate, &c., on the addition of ammonic chloride and ammonia. It is best separated from this precipitate by fusion with bisulphate of potash, as already described, but it must be remembered that the presence of much mineral acid prevents complete precipitation when the solution is boiled. Further, if phosphates are present, the precipitate will contain phosphoric oxide; it may be freed from this by fusion with sodium carbonate. A very good method of separating titanium from iron is to add tartaric acid and ammonia to the solution, and then precipitate the iron (as sulphide) with sulphuretted hydrogen. The filtrate contains the titanium, which is recovered by evaporating and igniting. It may be separated from zirconia by the action of sodium carbonate, which precipitates both; but when concentrated, redissolves the zirconia. The separation from large quantities of silica is best effected by evaporating with hydrofluoric acid, which volatilises the silicon; but sulphuric acid must be present, otherwise some titanium also will be lost, as may be seen from the following experiments,[77] in which oxide of titanium (pure, ignited) was evaporated to dryness with a quantity of hydrofluoric acid known by experiment to be sufficient to volatilise 1 gram of silica.
Without sulphuric acid, 0.0466 gram of titanic oxide left 0.0340 gram, showing a loss of about 25 per cent.
With sulphuric acid the following results were obtained:—
| Oxide taken. | Left after Evaporation and Ignition. |
| 0.0340 gram | 0.0340 gram |
| 0.0414 " | 0.0413 " |
| 0.0520 " | 0.0520 " |
| 0.0352 " | 0.0352 " |
The titanic hydrate thrown down by ammonia (or on boiling the solution from the bisulphate) is collected, washed, dried, ignited strongly with the addition of a little ammonic carbonate, and weighed. The substance is titanic oxide (TiO2), and is generally reported as such. It contains 60.98 per cent. of titanium. It should be white, if pure (Holland), white, yellow, or brown (Fresenius), or black (Tidy).[Pg 295]
A method has been proposed based on the reduction of titanic oxide by zinc in hydrochloric acid solutions to the sesquioxide. The reduction is marked by the development of a violet or green colour, the former with chlorides and the latter when fluorides are present. The quantity of titanium reduced is measured by titrating with permanganate of potassium solution. The water used must be free from dissolved oxygen.
Tungsten occurs in nature only in the oxidised state, or as tungstic acid (WO3), either free, as in wolframine, or combined with oxides of manganese and iron, as in wolfram, or with lime, as in scheelite. Wolfram occurs associated with tin ores, the value of which is consequently lowered. Both wolfram and scheelite are of considerable importance as a source of tungstic acid for the manufacture of sodium tungstate, which is used as a mordant and for some other purposes, and as a source of metallic tungsten, which is used in steel-making.
The tungsten minerals have a high specific gravity (6 to 7.5). On treatment with hydrochloric acid or aqua regia they are decomposed; the yellow tungstic acid separates and remains insoluble.
Tungsten itself is insoluble in nitric acid or aqua regia; but is converted into tungstic acid (WO3) by prolonged and strong ignition in air. Alloys containing tungsten leave tungstic acid after treatment with nitric acid or aqua regia. Tungstic acid may be got into solution after fusion with alkalies or alkaline carbonates. This solution gives with hydrochloric acid a white precipitate of tungstic acid, which becomes yellow on boiling, but the separation is not complete. Fusion with bisulphate of potash gives a residue, which does not dissolve in water, but is soluble in ammonic carbonate. For the assay of minerals containing tungsten these reactions are only occasionally taken advantage of for testing or purifying the separated tungstic acid.
Detection.—The minerals are easily recognised by their physical characters, and the yellow tungstic acid separated by boiling with acids is the best test for its presence; this, after decanting and washing, immediately dissolves in a few drops of dilute ammonia. A solution of tungstate acidulated with hydrochloric acid becomes intensely blue on the addition of stannous chloride and warming. Fused in a bead of microcosmic salt it[Pg 296] gives a clear blue colour (reddish-brown if iron is also present) in the reducing flame, but is colourless in the oxidising flame.
Solution and Separation.—The decomposition and solution of natural tungstates is difficult to effect owing to the separation of tungstic acid; the method of treatment is as follows:—Boil the finely-powdered substance with hydrochloric acid or aqua regia till it apparently ceases to be attacked; dilute, filter, and wash with dilute hydrochloric acid. Cover with dilute ammonia, and filter the solution, which contains ammonic tungstate, into an evaporating dish. Treat the residue again with acid, and again dissolve out the separated tungstic acid with ammonia, and repeat this operation until decomposition is complete. By this means there will be obtained—(1) a solution containing tungstate of ammonia; (2) an insoluble residue with silicates, and oxides of tin, niobium, tantalum, &c.; and (3) an acid solution containing the soluble bases. The tungstate of ammonia requires simple evaporation on the water-bath and gentle ignition in order to cause the tungstic acid to be left in an almost pure state; possibly, it may carry a little silica.
The tungstic acid is dissolved, and separated as ammonic tungstate, and, after evaporation, is gently ignited, the heat being increased towards the end. The residual tungstic acid is fixed, so that when the ammonia has been driven off it may be strongly heated without loss. It is a dark yellow or brown powder whilst hot, which becomes a light yellow on cooling. If any reduction has taken place it will be more or less greenish. It is weighed when cold, and is the trioxide or "tungstic acid" (WO3), which contains 79.31 per cent. of tungsten. After its weight has been taken its purity is checked by fusing with hydric potassic sulphate, extracting with water, and treating the residue with ammonic carbonate. Any silica present will be left undissolved; it should be separated and weighed, and its weight deducted from that of the tungstic acid found.
Determination of Tungstic Acid in Wolfram.—Take 2 grams of the finely-powdered sample and boil with 50 c.c. of hydrochloric acid for half an hour, adding 5 c.c. of nitric acid towards the end. Allow to stand overnight and boil again for 15 or 20 minutes; dilute with an equal volume of water, and filter. Wash with dilute hydrochloric acid, dissolve in a few c.c. of warm dilute ammonia, and dilute to 200 c.c. with distilled water; allow to settle, and filter. Evaporate in a weighed dish, ignite, and weigh.[Pg 297]
The following analysis will illustrate the composition of a sample of Cornish wolfram as brought into the market:—
| Tungstic acid | 50.1% |
| Cassiterite | 10.9 |
| Ferrous oxide | 24.6 |
| Manganous oxide | 5.4 |
| Niobic oxide, alumina, &c. | 3.5 |
| Silica | 1.2 |
| Copper oxide | 2.7 |
| Zinc oxide | 0.22 |
| Arsenic | 0.51 |
| Sulphur | 0.20 |
| ——— | |
| 99.33 |
These oxides are commonly met with in samples of wolfram and tinstone, especially niobic. They are probably present in the form of columbite, a niobate of iron and manganese; and tantalite, a tantalate of the same metals.
On boiling with hydrochloric acid they are both liberated, and remain for the greater part (all the niobic) in the insoluble residue with the tungstic acid. On removing the latter with dilute ammonia they remain as a white insoluble precipitate, very prone to run through the filter on washing. They may be dissolved in hydrofluoric acid either at once or after fusion with bisulphate of potash, and extraction with cold water. To the solution in hydrofluoric acid gradually add a boiling solution of acid potassium fluoride (HF, KF.). Potassic fluotantalate (soluble in 200 parts of water) separates out first, and afterwards potassic fluoniobate (soluble in 12 parts of water). The separated salts (after heating with sulphuric acid and washing out the potassium sulphate formed) are ignited with ammonic carbonate, and weighed as tantalic oxide (Ta2O5) and niobic oxide (Nb2O5) respectively.
They are both white powders. The oxide of niobium dissolved in a bead of microcosmic salt gives a bluish colour in the reducing flame. The oxide of tantalum dissolves in the bead, but gives no colour.
[76] This will give almost the whole of the tin; a further portion will be got in subsequent work, and must be added to this result.
[77] Published by P. Holland, in the Chemical News, vol. lix. p. 27.
Manganese occurs mainly as black oxide (MnO2) in the mineral pyrolusite; and, in a less pure form, in psilomelane and wad. The value of the ore depends rather on the percentage of available oxygen than on the proportion of metal present. The results of assays are generally reported as so much per cent. of the dioxide (MnO2). In smaller quantities it is very widely distributed. Manganese itself has a value for steel-making; or, rather, for the making of spiegeleisen and ferro-manganese, which are used in the Bessemer and Siemens processes. For this purpose the percentage of the metal (Mn) is required. Consequently the minerals of manganese may be considered in two aspects—(1) as a source of oxygen; and (2) as a source of manganese. These will require separate consideration.
The black oxide is mainly used in the preparation of chlorine, liberation of which it brings about when treated with hot hydrochloric acid, or with a mixture of common salt and sulphuric acid. The quantity of chlorine which is obtained depends upon the proportion of dioxide present;[78] and in assaying may either be measured by its equivalent of iodine liberated, or by the oxidising effect on an acid solution of ferrous sulphate. When the ore also carries substances which have a reducing effect (such as ferrous compounds), such assays will give, not the total dioxide (MnO2), but less, by the amount required to oxidise these impurities; and this is exactly what is required in valuing such an ore for commercial purposes. Manganese compounds are characterised by the readiness with which they may be converted into highly-oxidised bodies. Solution of manganese in hydrochloric acid, rendered alkaline with ammonia, yields a clear solution,[79] which rapidly takes up oxygen from the air, forming a brown precipitate of the oxide (Mn2O3). The addition of[Pg 299] bromine or chlorine to such a solution determines the precipitation of a still higher oxide (approximately MnO2). On treating a compound containing manganese with nitric acid and dioxide of lead (PbO2), the oxidation is carried still further, a purple-coloured solution of permanganic acid (HMnO4 or H2O.Mn2O7) being formed. On fusing minerals containing (even traces of) manganese with sodium carbonate in an open crucible, a green "melt" is obtained which owes its colour to sodium manganate (Na2MnO4 or Na2O.MnO3). This salt is soluble in water, forming a green solution; which, when rendered acid, rapidly changes into the permanganate with the characteristic purple colour. Permanganate of potash is a salt much used in assaying, with some properties of which the student will have already become familiar.
Compounds of manganese, on boiling with strong hydrochloric acid, yield manganous chloride[80] (MnCl2).
The properties given above serve for the detection of manganese; the higher oxides are distinguished by causing the evolution of chlorine (with its peculiarly suffocating smell) when acted on with hydrochloric acid; while the green "melt," with sodium carbonate, can be relied on for the recognition of manganese itself. There is no dry assay of manganese ores.
Strong hydrochloric acid is the best solvent for ores of manganese; but where the proportion of dioxide (MnO2) is required, the solution is effected during the assay. The ore should be in a very fine state of division before treatment with acids.
The separation of manganese from other metals is thus effected: Ignite, in order to destroy any organic matter which may be present; dissolve in hydrochloric acid, and evaporate to dryness, to separate silica. Take up with hydrochloric acid, dilute, pass sulphuretted hydrogen, and filter. Boil off the excess of gas, peroxidise the iron with a drop or two of nitric acid, and separate the iron as basic acetate (as described under Iron).[81] If the iron precipitate is bulky, it is dissolved in a little hydrochloric acid, reprecipitated, and the filtrate added to the original one. Neutralise with soda, and add bromine in excess; heat gradually to boiling, allow to settle, and filter. The precipitate is impure dioxide of manganese (containing alkalies and, possibly, cobalt or nickel).[Pg 300]
Dissolve the precipitate in hydrochloric acid, and boil; add a slight excess of carbonate of soda, warm, and filter. Wash with hot water, dry, carefully ignite in an open Berlin crucible, and weigh. The substance is the brown oxide (Mn3O4), and contains 72.05 per cent. of manganese. If the percentage of dioxide is required it may be calculated by multiplying the percentage of manganese by 1.582. It must be borne in mind that the manganese should never be calculated to dioxide except when it is known to exist in the ore only in that form.
The two methods are based on the oxidising effect of manganese dioxide; and if the metal does not already exist in this form it will require a preliminary treatment to convert it. The following method due to Mr. J. Pattinson[82] effects this: A quantity of the ore containing not more than .25 grams of the metal (Mn), is dissolved in hydrochloric acid in a pint beaker, and, if necessary, 3 or 4 c.c. of nitric acid are added to peroxidise the iron, and ferric chloride is added if required, so that there may be at least as much iron as manganese. Calcium carbonate is added till the solution is slightly red; and next the redness is removed by the cautious addition of acid; 30 c.c. of zinc chloride solution (containing 15 grams of zinc per litre) are added, the liquid is brought to boil and diluted to about 300 c.c. with boiling water; 60 c.c. of a solution of bleaching powder (33 grams to the litre and filtered), rendered slightly greenish by acid, are then run in and are followed by 3 grams of calcium carbonate suspended in 15 c.c. of boiling water. During effervescence the beaker is covered, the precipitate is stirred, and 2 c.c. of methylated spirit are mixed in. The precipitate is collected on a large filter, washed with cold water, and then with hot, till free from chlorine, which is tested for with starch and potassium iodide. The acid ferrous sulphate solution (presently described) is then measured into the beaker, and the precipitate, still in the paper, added; more acid is added (if necessary), and the solution is diluted and titrated. In place of bleaching powder solution, 90 c.c. of bromine water (containing 22 grams per litre) may be used.[Pg 301]
This method, which is the one commonly used, is based on the determination of the amount of ferrous iron oxidised by a known weight of the ore. It is known that 87 parts of the dioxide will oxidise 112 parts of ferrous iron;[83] therefore 1 gram will oxidise 1.287 gram of ferrous iron, or 1 gram of ferrous iron oxidised will be equivalent to 0.7768 gram of the dioxide. The finely-divided substance containing the dioxide is digested in a solution of a known quantity of iron in sulphuric acid. The iron, of course, must be in excess, which excess is determined when the ore is dissolved by titrating with standard permanganate or bichromate of potash solution. The assay resolves itself into one for the determination of ferrous iron, for which the standard solutions and method of working described under Iron are used.
The assay is as follows:—For rich ores, 2 grams of clean soft iron wire are treated, in a pint flask, with 100 c.c. of dilute sulphuric acid and warmed till dissolved. Carefully sample the ore, and in one portion determine the "moisture at 100° C.;" grind the rest in a Wedgwood mortar with a little pure alcohol until free from grit. This reduces the substance to a finely-divided state and assists solution. Evaporate off the alcohol and dry at 100° C., mix well, and keep in a weighing-bottle. Weigh up 2 grams and add them to the solution of iron in the flask; carefully wash it all down into the acid liquid. On rotating the flask the ore will rapidly dissolve, but gentle heat may be used towards the end to complete the solution. When the residue is clean and sandy-looking, and free from black particles, the flask is cooled, and the residual ferrous iron is determined by titration with "permanganate." The iron thus found, deducted from the 2 grams taken, will give the amount of iron peroxidised by the dioxide contained in the 2 grams of ore. This divided by 2 and multiplied by 77.68 will give the percentage of dioxide in the sample, or multiplied by 49.41 will give that of metallic manganese.
When the quantity of manganese or of the dioxide to be determined is small, it is not necessary to use 2 grams of iron; 1 gram, or even less, may be taken. The iron may be used in the form of a standard solution of ferrous sulphate and portions measured off, thus saving the labour of weighing.[Pg 302]
Determination of Dioxide in a Manganese Ore.—Weigh up 1 or 2 grams of the finely-powdered ore[84] and an equal weight of pure iron wire, dissolve the wire in 50 or 100 c.c. of dilute sulphuric acid, and, when solution is complete, add the ore and warm till it too is dissolved. Cool and titrate the remaining ferrous iron with the permanganate or bichromate of potassium solution.
For example, 0.7560 gram of pyrolusite and 1.000 gram of iron were taken and treated as above; 13.9 c.c. of "permanganate" (standard 100 c.c. = 0.4920 gram iron) were required; this indicates that 0.0684 gram of iron was left unoxidised by the ore. The iron oxidised, then, was 0.9316 gram (1.000 - 0.0684); multiplying this by 0.7768, we find that 0.7237 gram is the quantity of manganese dioxide which was present. This is equivalent to 95.77 per cent.;
0.7560 : 0.7237 :: 100 : 95.77.
It has been already stated that when dioxide of manganese is boiled with strong hydrochloric acid chlorine is given off, and that the amount of chlorine so liberated is a measure of the dioxide present. If the chlorine is passed into a solution of potassium iodide, an equivalent of iodine will be set free.[85] This is apparently a very indirect way of determining how much of the dioxide is present; but the reactions are very sharp, and the final determination of the iodine is an easy one.
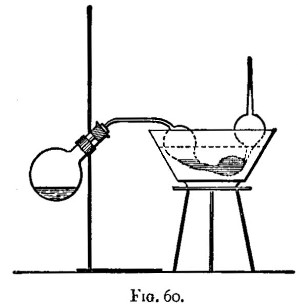
The finely-powdered sample of dioxide is placed in a small flask provided with an exit tube leading into a solution of potassic iodide (fig. 60). On adding hydrochloric acid and boiling, the chlorine evolved is driven into the iodide solution and there absorbed; the boiling is continued till the steam and hydrochloric acid fumes have driven the last portions of the chlorine out of the flask and into the solution. In this experiment there is a strong tendency for the iodide solution[Pg 303] to rush back into the flask. This tendency is overcome by avoiding draughts and regulating the heat; or by placing a lump of magnesite in the flask, which acts by evolving carbonic acid and so producing a steady outward pressure. When the distillation is finished the tube containing the iodine is detached and washed out into a beaker. If the solution is strongly acid it should be almost neutralised by the cautious addition of dilute ammonia. If crystals of iodine have separated, potassium iodide must be added in quantity sufficient to dissolve them. The condenser must be kept cool whilst the chlorine is passing into it.
The solution, transferred to a beaker, is titrated with a standard solution of sodic hyposulphite (100 c.c. = 1.27 gram iodine or 0.435 gram of dioxide of manganese). In titrating, the solution should be cold, or not warmer than 30° C. The bulk may vary from 100 to 200 c.c.; but it is best always to work with the same volume. The "hypo" is run in with constant agitation until the brown colour has been reduced to a light yellow; 5 c.c. of starch solution are then added and the titration cautiously continued until the end is reached; the finish is indicated by a change from blue to colourless.
The assay solution may be acidified with acetic, sulphuric, or hydrochloric acid before titrating with "hypo;" but it must be only faintly so. An excess of acid may be nearly neutralised with ammonia without interference, but excess of alkali is fatal. Bicarbonate of soda must not be used in excess; it is best to avoid it altogether. The assay solution should be titrated at once, as it weakens on standing; and the "hypo" solution should be standardised every two or three days, as its strength is not constant.
The standard solution of hyposulphite of soda is made by dissolving 25 grams of the salt (Na2S2O3.5H2O) in water and diluting to 1 litre. 100 c.c. are equivalent to 1.27 gram of iodine.
This solution is standardised by weighing, in a small beaker, about half a gram of iodine, to which is added a crystal or two of potassium iodide and a few drops of water. When dissolved, the solution is diluted to 100 c.c., and titrated in the manner described. The starch solution is made in the manner described under the iodide copper assay. 5 c.c. are used for each titration.
In determining the effects of variations in the condition of the assay a solution of iodine was used, which was equivalent in strength to the "hypo" solution. It was made by dissolving 12.7 grams of iodine with 25 grams of potassium iodide in a little water and diluting to 1 litre. 100 c.c. of this solution were found (at the time of the experiments) to be equivalent to 102.0 c.c. of the "hypo."[Pg 304]
Effect of Varying Temperature.—The bulk of the solution was 100 c.c.; 20 c.c. of iodine were taken, and 5 c.c. of starch solution were added towards the end as indicator. These conditions are also those of the other experiments, except where otherwise stated. Iodine being volatile, it is to be expected that with hot solutions low results will be obtained.
| Temperature | 15° | 20° | 40° | 60° | 80° |
| "Hypo" required | 20.4 c.c. | 20.4 c.c. | 20.1 c.c. | 19.2 c.c. | 15.5 c.c. |
These show that the temperature should not much exceed 20°.
Effect of Exposure of the Iodine Solution.—Twenty c.c. of the iodine were diluted to 100 c.c., and exposed for varying lengths of time in open beakers at the ordinary temperature, and then titrated.
| Time exposed | — | 1 day | 2 days | 3 days |
| "Hypo" required | 20.4 c.c. | 16.1 c.c. | 13.6 c.c. | 9.4 c.c. |
Effect of Varying Bulk.—These experiments were carried out in the usual way, bulk only varying.
| Bulk | 100.0 | c.c. | 200.0 | c.c. | 300.0 | c.c. | 500.0 | c.c. |
| "Hypo" required | 20.4 | " | 20.4 | " | 20.4 | " | 20.4 | " |
Effect of Varying Acid.—These experiments were under the usual conditions, the bulk being 100 c.c. The results were—
| Acetic acid | — | 1.5 | c.c. | 30.0 | c.c. |
| "Hypo" required | 20.4 c.c. | 20.7 | " | 20.7 | " |
| Hydrochloric acid | — | 1.5 | c.c. | 15.0 | c.c. |
| "Hypo" required | 20.4 c.c. | 20.6 | " | 20.9 | " |
| Sulphuric acid | — | 0.5 | c.c. | 20.0 | c.c. |
| "Hypo" required | 20.4 c.c. | 20.7 | " | 15.2 | "[86] |
| Nitric acid | — | 0.5 | c.c. | 10.0 | c.c. |
| "Hypo" required | 20.4 c.c. | 21.5 | " | could not be titrated. |
In the application of this titration to the assay of manganese ores, hydrochloric and hydriodic acids are the only ones likely to be present.
Effect of Alkalies.—On theoretical grounds the presence of these is known to be inadmissible. A solution rendered faintly alkaline with ammonia required only 11.2 c.c. of "hypo;" and another, with 0.5 gram of caustic soda, required 4.0 c.c. instead of 20.4 c.c. as in neutral solutions.
Effect of nearly Neutralising Hydrochloric Acid Solutions with Ammonia.—Provided care is taken not to add[Pg 305] excess of ammonia, this has a good effect, counteracting the interference of excess of acid. Thus 20 c.c. of iodine (as before) required 20.4 c.c. of "hypo;" with 15 c.c. of hydrochloric acid 20.7 c.c. were required, but with 15 c.c. of acid, nearly neutralised with dilute ammonia 20.4 c.c. were used.
Effect of the Addition of Starch.—The addition of varying quantities of starch has no effect, provided it is added when the titration is nearly finished, as the following experiments show:—
| Starch added | 1.0 | c.c. | 5.0 | c.c. | 10.0 | c.c. | 50.0 | c.c. |
| "Hypo" required | 20.4 | " | 20.4 | " | 20.4 | " | 20.5 | " |
But if the starch is added before the titration, the results are liable to error.
| Starch added | 1.0 | c.c. | 50.0 | c.c. |
| "Hypo" required | 20.4 | " | 24.0 | " |
The starch should be used fresh, and is best made on the day it is used; after four days the finishing point is not so good.
Effect of Varying Potassium Iodide.—An excess of iodide is always required to keep the iodine in solution; a larger excess has little effect.
| Iodide added | — | 1 gram | 20 grams |
| "Hypo" required | 20.4 c.c. | 20.5 c.c. | 20.6 c.c. |
The 20 c.c. of iodine used, itself contained 0.5 gram of potassium iodide.
Effect of Foreign Salts.—
| Bicarbonate of soda added | — | 0.5 gram | 1.5 gram | 5.0 grams |
| "Hypo" required | 20.4 c.c. | 18.2 c.c. | 17.1 c.c. | 16.0 c.c. |
The solution obviously must be free from bicarbonate of soda. This should be remembered, since when titrating arsenic assays with iodine it must be present; and students must avoid confounding the two titrations.
In some other experiments, in which 10 grams each of the salts were taken, the following results were obtained:—
| Salt added | — | AmCl | AmNO3 | Am2SO4 |
| "Hypo" required | 20.4 c.c. | 20.5 c.c. | 20.3 c.c. | 20.2 c.c. |
| Salt added | NaCl | NaNO3 | Na2SO4 |
| "Hypo" required | 20.3 c.c. | 20.4 c.c. | 20.4 c.c. |
Effect of Varying Iodine.—
| Iodine added | 1.0 | c.c. | 10.0 | c.c. | 20.0 | c.c. | 50.0 | c.c. | 100.0 | c.c. |
| "Hypo" required | 1.3 | " | 10.2 | " | 20.4 | " | 51.0 | " | 102.0 | " |
Determination of Dioxide in a Manganese Ore.—Weigh up 0.25 to 0.3 gram of the powdered ore; place in a flask, cover[Pg 306] with 10 c.c. of hydrochloric acid, and close the flask with a paraffined cork, and bulbs (as shown in fig. 60), having previously charged the bulb with 5 grams of potassium iodide in strong solution. Heat the flask, and boil cautiously for about fifteen minutes. Wash the contents of the bulbs into a large beaker, nearly (but not quite) neutralise with dilute ammonia, and titrate with the standard "hypo."
As an example, 0.2675 gram of pyrolusite was taken, and required 60.3 c.c. of standard "hypo" (100 c.c. equal 1.185 gram iodine, or 0.4042 gram MnO2), which equals 0.2437 gram of the dioxide or 91.1 per cent.
When compounds of manganese free from chlorides are boiled with nitric acid and dioxide of lead,[87] the manganese is converted into permanganic acid, which is soluble and tints the solution violet. The depth of colour depends on the amount of manganese present, and this should not much exceed 10 milligrams. A quantity of substance containing not more than this amount of manganese should be boiled for a few minutes with 25 c.c. of a solution containing 5 c.c. of nitric acid, and 10 or 20 c.c. of dilute sulphuric acid, with 2 or 3 grams of lead dioxide. Filter through asbestos, wash by decantation with dilute sulphuric acid, make up with distilled water[88] to a definite bulk, and take a measured portion for the colorimetric determination.
The standard solution of manganese is made by dissolving 0.1435 gram of permanganate of potash (KMnO4) in a little water acidulated with nitric acid, and diluting to 1 litre. One c.c. will contain 0.05 milligram of manganese.
1. What percentage of manganese (Mn) is contained in permanganate of potash (KMnO4)?
2. Ten c.c. of a solution of permanganate of potash is found to oxidise 10 c.c. of an acid solution of ferrous sulphate. The manganese is determined in the titrated solution by precipitation as dioxide and titrating. How much of the ferrous solution will be oxidised in the second titration?
3. What weight of potassium iodide would be just sufficient to absorb the chlorine evolved by 0.5 gram of pure dioxide of manganese?[Pg 307]
4. What weight of iron must be dissolved up so as to have an excess of 0.25 gram after oxidation by 1 gram of pure dioxide?
5. What weight of the brown oxide, Mn2O4 will be left on igniting 1 gram of the pure dioxide?
Chromium occurs in nature chiefly as chromite or chrome iron ore (FeO2Cr2O3, with more or less MgO and Al2O3), which is the chief ore. It is a constituent of some silicates, and is frequently met with in very small quantities in iron ores. It occurs as chromate in crocoisite (PbCrO4), and some other rare minerals.
The metal is used in steel-making. Steel containing about 0.5 per cent. of it is rendered very hard; but its chief value is in its salts, the chromates. These are highly-coloured compounds, generally red or yellow. Some of the insoluble chromates are used as pigments; chromate of lead or chrome-yellow is the most important. The soluble chromates, those of soda and potash, are valuable chemicals, and are largely used in the preparation of pigments, dyeing and tanning, and as oxidising agents.
Chromium forms two important classes of compounds—chromic salts, corresponding to the oxide Cr2O3, and chromates, which contain the trioxide CrO3. Solutions of chromic salts are green, whilst those of the chromates are yellow. Chromates are reduced to chromic salts by the action of most reducing agents in the presence of an acid; and this property is used in assaying for the volumetric determination of ferrous iron, &c. The chromates in solution are more stable than other similar oxidising agents, and consequently are generally used in the laboratory as one of the standard oxidising agents for volumetric analysis. They have the disadvantage of requiring an outside indicator. Bichromate of potash (K2Cr2O7) is the salt generally used for this purpose.
Chromic salts are oxidised to chromate by fusion with "fusion mixture" and nitre, or by treating with chlorine in an alkaline solution.
Chromic salts closely resemble those of ferric iron, and in the ordinary course of analysis chromic hydrate (green) is precipitated together with ferric hydrate, alumina, &c., on the addition of ammonic chloride and ammonia. The ignited oxide, Cr2O3, however, is not reduced on heating to redness in a current of hydrogen.
Detection.—Chromium is detected by fusing the powdered substance with "fusion mixture" and nitre. The melt is extracted with water and filtered. The filtrate is acidified with acetic acid, and treated with a few drops of a solution of lead[Pg 308] acetate. A yellow precipitate indicates chromium. Substances containing chromium impart a green colour to the borax bead in both flames. Small quantities of chromate in neutral solution can be found by the dark or violet-red colouration imparted thereto on boiling with a dilute decoction of logwood.
Solution and Separation.—Chromates and chromic salts are generally soluble in water or dilute acids. Chrome iron ore, however, and ignited chromic oxide are insoluble; and the former presents considerable difficulty on attempting to open up by the usual methods. A large number of mixtures have been tried in order to get all the chromium in a soluble form. Among these are the following. One part of the very finely-powdered ore is fused with any of these mixtures.
(1) 10 parts of bisulphate of potash.
(2) 5 parts of bisulphate of potash and 5 parts of potassium fluoride.
(3) 5 parts of hydric potassic fluoride.
(4) 12 parts of bisulphate of potash; and, afterwards, with 6 parts of
carbonate of soda and 6 parts of nitre.
(5) 8 parts of borax; afterwards, with carbonate of soda till it ceases
to effervesce; then, with 3 parts of carbonate of soda and 3 of
nitre.
(6) 4 parts of borax and 6 parts of fusion mixture.
(7) 12 parts of caustic potash.
(8) 10 parts of caustic soda and 30 of magnesia.
(9) 5 parts of caustic soda and 3 of magnesia.
(10) 2 parts of carbonate of soda and 1 of lime.
(11) 6 parts of soda-lime and 2 of chlorate of potash.
(12) Sodium peroxide.
Of these, numbers 1, 2, and 3 yield the chromium in a form soluble in dilute acids, as chromic salt. The rest in a form soluble in water, as potassium or sodium chromate.
On boiling an insoluble chromium compound with chlorate of potash and nitric acid, the chromium passes into solution as chromate. This method, however, does not answer for chrome iron ore. In the fusion methods the ore must be very finely powdered, well mixed with the fluxes, and subjected to a prolonged fusion in a platinum vessel at a high temperature. Undecomposed particles require re-fusion.
The aqueous extract containing the chromate is ready for volumetric work, except in those cases where nitre has been used. For gravimetric work the solution is acidified with hydrochloric acid, then mixed with ammonia in slight excess, boiled, and filtered. The filtrate is acidified with hydrochloric acid, and treated with sulphuretted hydrogen, warmed, rendered slightly alkaline with ammonia, and the gas again passed. The chromium is precipitated as chromic hydrate mixed with sulphur from the[Pg 309] reduction with sulphuretted hydrogen. It is filtered off, washed with hot water, and ignited. It is weighed as chromic oxide.
The solution containing the chromium, freed from other metals and earths and in the form of (green) chromic salt, is heated to boiling. If any chromate is present reduce it with sodium sulphite or sulphuretted hydrogen. Add ammonia in slight excess, boil till the liquid is free from a red tint, and allow to settle for a few minutes. Filter, wash with hot water, dry, and ignite strongly in a loosely-covered crucible. Cool, and weigh. The substance is chromic oxide, Cr2O3, and contains 68.62 per cent. of chromium. It is a dark-green powder insoluble in acids.
When, as is generally the case, the chromium exists altogether as chromate (phosphates and arsenates being absent) it is best to proceed as follows:—Render the solution acid with acetic acid, then add sodium acetate to the solution and heat nearly to boiling; next treat with a slight excess of acetate of lead, and boil. Allow to settle, and filter. Wash the precipitate with hot water, dry in the water-oven or at a low temperature. Transfer the precipitate to a weighed Berlin crucible, burn the filter separately, ignite below redness, cool in the desiccator, and weigh. The substance is lead chromate, PbCrO4, and contains 16.1 per cent. of chromium, or 23.53 per cent. of chromic oxide (Cr2O3).
This is based on the oxidation of ferrous iron by the solution containing the chromium as chromate. A known weight of iron (0.5, 1, or 1.5 gram, according to the quantity of chromate) is dissolved in 50 c.c. of dilute sulphuric acid. The solution containing the chromate is added, and the remaining ferrous iron titrated with the permanganate or bichromate of potassium solution, as described under Iron. The iron thus found is deducted from that taken, and the difference gives the iron oxidised by the chromate. This multiplied by 0.3101 gives the chromium, Cr, and when multiplied by 0.4529 gives the chromic oxide, Cr2O3.
Small quantities of chromium may be determined, after conversion into chromate, colorimetrically. The solution, which should not contain more than a few milligrams in 100 c.c., is acidified with acetic acid and compared against an equal volume[Pg 310] of water rendered acid with acetic acid and tinted with a standard bichromate of potassium solution. This standard bichromate is made by dissolving 2.827 grams of the salt in water and diluting to 1 litre. One c.c. will contain 1 milligram of chromium, Cr. The manner of working this assay is the same as that adopted in the other colorimetric processes.
Determination of Chromium in Steel.[89]—Weigh up 2.4 grams, dissolve in hydrochloric acid, and evaporate to dryness. Fuse with sodium carbonate and nitre, extract with water, and make up to 301 c.c. Take 250 c.c. of the clear liquor, boil with hydrochloric acid, add sodium phosphate, and then ammonia in slight excess. Heat till clear. Filter off the precipitate, dissolve it in hydrochloric acid, and evaporate to dryness. Take up with a little acid, filter, and precipitate with a slight excess of ammonia. Wash, ignite, and weigh as chromium phosphate (3Cr2O3,2P2O5), which contains 42.2 per cent. of chromium.
Vanadium occurs in certain rare minerals, such as vanadinite (3Pb3(VO4)2.PbCl2), a vanadate of lead; mottramite, a vanadate of copper and lead; and dechenite, a vanadate of lead and zinc. It is occasionally found in iron and copper ores and in the slags from them. In Spanish copper-precipitates it is found along with chromium, and is probably derived from the iron used for precipitating. The vanadates, like the chromates, are coloured compounds, generally yellow or red. On reduction, blue solutions are got. In their general reactions vanadates resemble phosphates.
Vanadium is detected by the red colouration produced by passing sulphuretted hydrogen into ammoniacal solutions for some time. On adding an acid to the filtered solution a brown precipitate of the sulphide is produced. This gives with borax a colourless bead in the oxidising, and a green one in the reducing, flame.
It is separated by fusing the ore with potassic nitrate, extracting with water and precipitating with baric chloride. The precipitate is boiled with dilute sulphuric acid, filtered, neutralised with ammonia, and saturated with ammonic chloride. Ammonium vanadate separates out. It is filtered off, ignited, and weighed as vanadic oxide, V2O5, containing 56.18 per cent. of vanadium.[Pg 311]
Molybdenum occurs in nature chiefly as molybdenite (MoS2); it also occurs in wulfenite, a molybdate of lead (PbMoO4), and in molybdic ochre (MoO3).
Molybdate of ammonia is an important reagent in the determination of phosphates, the manufacture of which compound is the chief purpose to which molybdenum is applied.
Iron and copper ores frequently contain molybdenum, sometimes in quantity; consequently it is met with in slags and pig-iron.
Molybdenum forms several series of salts. In those corresponding to the lower oxides it is basic; but the trioxide (MoO3) is the acid oxide which forms a series of salts called the molybdates. All molybdenum compounds are converted into the trioxide by boiling with nitric acid. The trioxide is a white powder readily dissolved by ammonia. It fuses at a red heat, and volatilises freely in contact with air. It is slightly soluble in water.
Molybdates are easily reduced, with the production of coloured solutions, by most reducing agents. Sulphuretted hydrogen first produces a blue tint, and then precipitates a brown sulphide. The precipitation as sulphide is only complete on prolonged treatment; a green colour indicates that some molybdenum still remains in solution. The precipitated sulphide is soluble in ammonium sulphide.
Detection.—Molybdenum is detected by its behaviour with sulphuretted hydrogen. Molybdenite can only be mistaken for graphite, from which it is easily distinguished by yielding sulphur dioxide on roasting, and by giving, on charcoal, a yellowish white incrustation, which becomes blue on touching it for a moment with the reducing flame. The borax-bead is colourless in the oxidising, and dark-brown in the reducing, flame.
The solution containing the molybdate is neutralised and treated with an excess of mercurous nitrate. The precipitate is allowed to settle for some time, filtered, and washed with a dilute solution of mercurous nitrate. Then it is dried, transferred to a weighed Berlin crucible containing ignited oxide of lead, mixed, ignited, and weighed. The increase in weight gives the amount of trioxide, MoO3. This contains 66.7 per cent. of molybdenum.[Pg 312]
Uranium occurs chiefly as pitchblende, which is an impure oxide (U3O8). It is also found as sulphate in uranochre, johannite, &c.; and as phosphate in the uranites, torbernite (hydrated phosphate of uranium and copper), and autunite (hydrated phosphate of uranium and lime). It also occurs in some rarer minerals.
The oxide is used for colouring glass; and the nitrate and acetate are used as reagents. "Uranium yellow," used for enamel painting, is sodium uranate. The uranates, in which the oxide of uranium acts as an acid, are mostly insoluble and of secondary importance.
Uranium forms two families of salts, uranous and uranic; corresponding to the oxides UO2 and UO3 respectively. The former are generally green and the latter yellow. Uranous salts are converted into uranic by boiling with nitric acid or other oxidising agents. Uranic salts, on the other hand, are easily reduced by sulphuretted hydrogen, stannous chloride or zinc. This property is made use of in determining the quantity of uranium in pure solutions by titrating with permanganate of potassium solution as in the case with iron.
Detection.—The most characteristic reaction of the uranium compounds is their behaviour in the presence of alkaline carbonates in which they are freely soluble; even ammonium sulphide will not precipitate uranium from these solutions. On neutralising the carbonate with an acid a uranate of the alkali is precipitated. Ammonia or sodic hydrate (free from carbonates) give yellow precipitates, which are insoluble in excess of the reagent, but are soluble in acids. Ferrocyanide of potassium gives a reddish-brown precipitate. Uranium colours the borax-bead yellowish-green in the oxidising, and green in the reducing, flame.
Solution and Separation.—The compounds of uranium are soluble in acids. Powder the substance and evaporate with an excess of nitric acid. Take up with hydrochloric acid, dilute, pass sulphuretted hydrogen, and filter. Peroxidise the filtrate with a little nitric acid, add an excess of ammonic carbonate and some ammonium sulphide, and filter. Render the solution acid, boil; and precipitate the uranium by means of ammonia. Filter off, and wash it with dilute ammonic chloride. Ignite, and weigh [Pg 313]as protosesqui-oxide, U3O8.
The solution containing the uranium free from other metals is, if required, first peroxidised by boiling with nitric acid. Ammonia in slight excess is added to the nearly-boiling solution. A yellow precipitate is formed, which is filtered off hot and washed with a dilute solution of ammonium chloride. The precipitate is dried and ignited; and weighed as U3O8, which contains 84.8 per cent. of uranium.
This is based on the precipitation of uranium as phosphate from acetic acid solutions and the recognition of complete precipitation by testing with potassic ferrocyanide; it is the converse of the process for the volumetric determination of phosphate.
The standard solution of phosphate is prepared by dissolving 29.835 grams of hydric sodic phosphate (Na2HPO4.12H2O) in water and diluting to 1 litre. 100 c.c. will be equivalent to 2 grams of uranium.
Take 1 gram of the sample (or, if poor in uranium, 2 grams) and separate the uranium as described. Dissolve the precipitate in nitric acid and evaporate to a small bulk, add 2 grams of sodium acetate, dilute with water to 100 c.c., and boil. Titrate the boiling solution with the sodium phosphate till it ceases to give a brown colouration with potassium ferrocyanide. Calculate the percentage in the usual way.
[78] MnO2 + 4HCl = MnCl2 + Cl2 + 2H2O.
[79] Provided a sufficiency of ammonic chloride is present.
[80] With some silicates, &c., a preliminary fusion with sodium carbonate will be necessary.
[81] Instead of sodium acetate, ammonium succinate can be used.
[82] Journ. Soc. Chem. Industry, vol. x. p. 333.
[83] MnO2 + 2FeSO4 + 2H2SO4 = Fe2(SO4)3 + MnSO4 + 2H2O.
[84] If the ore is very rich, a smaller quantity (0.75 or 1.5 gram) must be taken; otherwise the iron will be insufficient.
[85]
MnO2 + 4HCl = MnCl2 + 2H2O + Cl2.
Cl2 + 2KI = 2KCl + I2.
[86] Iodine probably lost by volatilisation.
[87] Obtained as a brown powder by digesting red lead with nitric acid and filtering.
[88] The water for dilution and the dilute sulphuric acid used for washing should be previously tested, to see they have no reducing action, with dilute permanganate of potassium solution.
[89] Arnold and Hardy, Chemical News, vol. lvii. p. 153.
Alumina, the oxide of aluminium (Al2O3), is found in nature fairly pure in the mineral corundum; transparent and coloured varieties of which form the gems sapphire and ruby. A coarser compact variety contaminated with oxide of iron constitutes emery. Compounded with silica, alumina forms the base of clays and many rock-forming minerals. China clay (or kaolin) is used as a source of alumina. Bauxite, hydrated alumina, is also used for the same purpose—that is, for the preparation of sulphate of alumina. The mineral cryolite is a fluoride of aluminium and sodium.
Corundum is characterised by a high specific gravity (4.0) and extreme hardness. By these it is distinguished from felspar and similar minerals, which it somewhat resembles in general appearance.
Aluminium is used for a variety of small purposes: it is white, light, and very tenacious; but owing to the difficulty of its reduction it is expensive.
Aluminium forms one series of salts which closely resemble those of ferric iron. It forms an interesting series of double sulphates, known as the alums. Common potash alum is Al2(SO4)3,K2SO4,24H2O.
Detection.—Alumina is not precipitated from its acid solution by sulphuretted hydrogen, but it is thrown down by ammonia (with the other earths) as a white hydrate, soluble in soda and insoluble in ammonic carbonate. Filtered off and ignited, it assumes, after treatment with nitrate of cobalt before the blowpipe, a blue colour which is characteristic. With natural compounds containing metallic oxides this colour is masked. It is more satisfactory to make a separation in the wet way and to test the ignited oxide.
Separation and Solution.—If the substance is insoluble in hydrochloric acid it is finely powdered and fused with "fusion mixture" with the help, in the case of corundum (which is very refractory) of a little caustic soda or potash. The method of[Pg 315] working is the same as that described for opening up silicates. See under Silica. Corundum cannot be powdered in Wedgwood, or even agate, mortars; since it rapidly wears these away and becomes contaminated with their powder. It is best to use a hard steel mortar and to extract the metallic particles from the bruised sample with a magnet or dilute acid.
When the substance has been completely attacked and dissolved, it is evaporated to dryness with an excess of hydrochloric acid on the water-bath to render any silica present insoluble. The residue is extracted with hydrochloric acid and freed from the second group of metals by means of sulphuretted hydrogen. The filtrate from this (after removing the sulphuretted hydrogen by boiling) is nearly neutralised, and treated with 8 or 10 grams of hyposulphite of soda[90] in solution. It is then boiled till the sulphurous oxide is driven off. The precipitate is filtered off, ignited, and weighed as alumina.
It is sometimes more convenient to proceed as follows:—After boiling off the sulphuretted hydrogen peroxidise the iron with a little nitric acid, add a solution of ammonic chloride, and then ammonia in very slight excess; boil, filter, wash, ignite, and weigh the oxides. These generally consist of ferric oxide and alumina. It is a common practice to determine the iron, calculate it to ferric oxide, and so to estimate the alumina indirectly. This may be done either by igniting in a current of hydrogen and estimating the iron by the weight of oxygen lost; or, by dissolving with sulphuric and hydrochloric acids, and determining the iron volumetrically. It should be borne in mind that these oxides will also contain any phosphoric oxide that happened to be in the mineral.
In general analyses of samples containing alumina, it may be contained in both the soluble and insoluble portions. In these cases it is better to fuse the sample with "fusion mixture" before treatment with acids. The alumina in the fused mass will exist in a state soluble in acids.
Solutions containing alumina free from the other metals are diluted to a convenient bulk and heated nearly to boiling. Add chloride of ammonium, and then ammonia in slight excess; boil, allow to settle, filter, and wash with hot water. Dry the precipitate, and ignite in a platinum or porcelain crucible at the strongest heat. Cool, and weigh. The substance [Pg 316]is alumina, Al2O3, which contains 52.94 per cent. of aluminium. It is only in special cases, such as the analysis of metals and alloys, that it is reported as aluminium. The percentage of alumina is generally given.
Ignited alumina is difficultly soluble in acids; it is not reduced by hydrogen at a red heat. Ignited with ammonium chloride portions are volatilised.
Direct Determination of Alumina in the Presence of Iron.—The iron and alumina are precipitated as hydrates by ammonia. The precipitate is dissolved in hydrochloric acid and the iron reduced to the ferrous state. It is then added to a hot solution of potash or soda. The solution is boiled till the precipitate settles readily, filtered, and washed with hot water. The alumina is contained in the filtrate, which is acidified with hydrochloric acid and the alumina precipitated therefrom as hydrate with ammonia, as just described.
Determination of Alumina in the Presence of Phosphates and Iron.—For details, see a paper by R.T. Thomson in the "Journal of the Society of Chemical Industry," v. p. 152. The principles of the method are as follows:—If the substance does not already contain sufficient phosphoric oxide to saturate the alumina, some phosphate is added. The iron is reduced to the ferrous state and phosphate of alumina precipitated in an acetic acid solution. It is purified by reprecipitation, ignited, and weighed as phosphate (Al2O3,P2O5), which contains 41.8 per cent. of alumina, Al2O3.
Moisture.—Take 5 grams of the carefully-prepared sample and dry in the water-oven till the weight is constant.
Loss on Ignition.—Weigh up 2 grams of the sample used for the moisture determination, and ignite in a platinum-crucible to redness, cool, and weigh.
Silica and Insoluble Silicates.—Weigh up another 2 grams of the dried sample, and place them in a platinum dish; moisten with water, and cover with 20 c.c. of sulphuric acid. Evaporate and heat gently to drive off the greater portion of the free acid. Allow to cool; and repeat the operation. Extract by boiling with dilute hydrochloric acid, filter, wash, dry, ignite, and weigh. The quantity of insoluble silicates is determined by dissolving out the separated silica with a strong boiling solution of sodium carbonate. The residue (washed, dried, and ignited) is weighed, and reported as "sand."
Alumina and Ferrous Oxide.—To the filtrate from the silica add "soda" solution till nearly neutral, and then sodium[Pg 317] acetate. Boil and filter off the precipitate. Reserve the filtrate. Dissolve the precipitate in hydrochloric acid, and dilute to exactly 200 c.c. Divide into two parts of 100 c.c. each. In one determine the iron by reducing and titrating in the way described under volumetric iron. Calculate the percentage as ferrous oxide, unless there are reasons to the contrary, also calculate its weight as ferric oxide. To the other portion add ammonia in slight excess, and boil. Filter, wash with hot water, dry, ignite, and weigh as mixed alumina and ferric oxide. The weight of the ferric oxide has already been determined in the first portion: deduct it, and the difference is the weight of alumina.
Lime.—To the reserved filtrate, concentrated by evaporation, add ammonium oxalate and ammonia; boil, filter, ignite strongly, and weigh as lime.
Magnesia is separated from the filtrate by adding sodium phosphate. It is weighed as magnesium pyrophosphate.
Potash and Soda.—These are determined in a fresh portion of the sample by Lawrence Smith's method, as described on page 333.
This is an oxide of thorium, ThO2. It is only found in a few rare minerals. It is a heavy oxide, having, when strongly ignited, a specific gravity of 9.2. In the ordinary course of analysis it will be separated and weighed as alumina. It is separated from this and other earths by the following method. The solution in hydrochloric acid is nearly neutralised and then boiled with sodium hyposulphite. The thoria will be in the precipitate. It is dissolved, and the solution heated with ammonium oxalate in excess. The precipitate is thorium oxalate, which is washed with hot water, dried, and ignited. It is then weighed as thoria, ThO2. Thoria which has been ignited is not readily soluble in acids.
The oxide of zirconium, ZrO2, is found in the mineral zircon, a silicate of zirconia, ZrSiO4. When heated intensely it becomes very luminous, and is used on this account for incandescent lights.
In the ordinary course it is thrown down by ammonia with the other earths, from which it is thus separated:—The hydrates precipitated in the cold, and washed with cold water, are dissolved[Pg 318] in hydrochloric acid, nearly neutralised with soda, and precipitated by boiling with hyposulphite of soda. Dissolve; and from the hydrochloric acid solution precipitate the thoria (if any) with ammonium oxalate. To the filtrate add carbonate of ammonia, which will precipitate any titanium present. The zirconia will be in solution, and is recovered by precipitating with potassium sulphate, or by evaporating the solution and igniting. It is separated from alumina by taking advantage of its insolubility in potassic hydrate.
It is estimated in zircons in the following way:—The powdered substance is fused with bisulphate of potash, and extracted with dilute sulphuric acid. The residue is fused with caustic soda and extracted with water. The portion not dissolved, consisting of zirconate of soda, is dissolved in hydrochloric acid. The solution is diluted, filtered if necessary, and treated with ammonia in excess. The precipitate is filtered off, washed with hot water, dried, ignited, and weighed as zirconia, ZrO2. This is a white powder, which is insoluble in acids; even in hydrofluoric acid it is only slightly attacked.
Cerium occurs as silicate (together with the oxides of lanthanum, didymium, iron and calcium) in the mineral cerite, which is its chief source. It also occurs as phosphate in monazite, and as fluoride in fluocerite. The oxalate is used in medicine. Cerium forms two classes of salts corresponding to the oxides, cerous oxide (Ce2O3) and ceric oxide (CeO2). Compounds of cerium with volatile acids yield dioxide on ignition; and this, on solution in hydrochloric acid, yields cerous chloride and chlorine.
In the ordinary course cerium is thrown down along with alumina and the other earths by ammonia. It is separated by dissolving the hydrates in hydrochloric acid, and oxidizing with chlorine water. On treating with oxalic acid, cerium, lanthanum, and didymium are precipitated as oxalates, which on ignition are converted into oxides. These are soluble in acids. Their solution in hydrochloric acid is nearly neutralised; acetate of soda is then added, and an excess of sodium hypochlorite. On boiling, the cerium is precipitated as dioxide, which is filtered off, ignited, and weighed.
Cerium is detected by giving with borax a bead which is yellow in the oxidising, and colourless in the reducing flame. Traces of cerium compounds boiled with dioxide of lead and nitric acid will give a yellow solution.[Pg 319]
occur together with cerium in cerite, and are separated with that metal as oxalates, as described under Cerium.
Didymium salts have a rose or violet colour, and impart (when in sufficient quantity) the same colour to the borax bead. Solutions have a characteristic absorption-spectrum.
The separation of lanthanum and didymium in the solution from which the cerium has been precipitated is effected by precipitating them together as oxalates, igniting, and dissolving in dilute nitric acid. This solution is then evaporated to dryness and ignited, for a few minutes, just below redness. A subnitrate of didymium is formed, and remains as an insoluble residue on extracting with hot water. The separated salts are treated with ammonia and ignited, and weighed as oxides (La2O3 and Di2O3).
Yttria is found in gadolinite and some other rare minerals. It is precipitated along with the other earths by ammonia. It is distinguished by the insolubility of its hydrate in potash, by the insolubility of its oxalate in oxalic acid, and by not being precipitated by hyposulphite of soda or potassium sulphate. Further, it is precipitated by potash in the presence of tartaric acid as an insoluble tartrate. This reaction distinguishes the members of the yttria group from most of the other earths. The other members of the group closely resemble it, and amongst them are erbia, terbia, ytterbia, scandia, &c.
The oxide of beryllium, BeO (also known as glucina), occurs in nature mainly as silicate. Beryl, the green transparent variety of which is the emerald, is the best known of these. It is a silicate of alumina and beryllia.[91] Some other minerals in which it occurs are phenakite, euclase, and chrysoberyl.
In the ordinary course of analysis, beryllia will be precipitated with alumina, &c., by ammonic hydrate. It is distinguished by the solubility of its hydrate in ammonic carbonate, by not being precipitated by boiling with sodium hyposulphite, and by not being precipitated by ammonic sulphide from an ammonic carbonate solution.
The analysis of silicates containing beryllia is thus effected.[Pg 320] The finely powdered substance is fused with twice its weight of potassium carbonate; and the "melt" is extracted with water, and evaporated with a slight excess of sulphuric acid to render the silica insoluble. Treat with water, filter, and evaporate the filtrate until a crust is formed. Potash alum crystallises out. The liquor is poured off into a warm strong solution of ammonium carbonate. Ferric hydrate and alumina will be precipitated. They are filtered off, re-dissolved, and again precipitated in ammonic carbonate solution; the combined filtrates are boiled for some time, and acidified slightly with hydrochloric acid. The carbon dioxide is boiled off, and the beryllia is then precipitated as hydrate with ammonia. The hydrate is washed with hot water, dried, ignited, and weighed as beryllia, BeO.
Beryllia has a specific gravity of 3.08. It is white, infusible, and insoluble in water. After ignition, it is insoluble in acids, except sulphuric, but is rendered soluble by fusion with alkalies.
Beryllia, in a solution of carbonate of ammonia, is precipitated as carbonate on boiling in proportion as the carbonate of ammonia is volatilised. The hydrate is dissolved by a boiling solution of ammonic chloride, ammonia being evolved.
Lime is an oxide of calcium, CaO. It occurs abundantly in nature, but only in a state of combination. The carbonate (CaCO3), found as limestone, chalk, and other rocks, and as the minerals calcite and arragonite, is the most commonly occurring compound. The hydrated sulphate, gypsum (CaSO4.2H2O), is common, and is used in making "plaster of Paris." Anhydrite (CaSO4) also occurs in rock masses, and is often associated with rock salt. Phosphate of lime, in the forms of apatite, phosphorite, coprolite, &c., is largely mined. Lime is a component of most natural silicates. Calcium also occurs, combined with fluorine, in the mineral fluor (CaF2). In most of these the acid is the important part of the mineral; it is only the carbonate which is used as a source of lime.
Lime, in addition to its use in mortars and cements, is valuable as a flux in metallurgical operations, and as a base in chemical work on a large scale. A mixture of lime and magnesia is used in the manufacture of basic fire-bricks.
Carbonate of lime on ignition, especially when in contact with reducing substances, loses carbonic acid, and becomes lime. This[Pg 321] is known as "quicklime"; on treatment with water it becomes hot, expands, and falls to a powder of "slaked lime" or calcium hydrate (CaH2O2). The hydrate is slightly soluble in water (0.1368 gram in 100 c.c.), forming an alkaline solution known as lime-water. Calcium hydrate is more generally used suspended in water as "milk of lime."
As a flux it is used either as limestone or as quicklime. Silica forms with lime a compound, calcium silicate, which is not very fusible; but when alumina and other oxides are present, as in clays and in most rocky substances, the addition of lime gives a very fusible slag.
Detection.—Calcium is detected by the reddish colour which its salts impart to the flame. It is best to moisten with hydrochloric acid (or, in the case of some silicates, to treat with ammonium fluoride) before bringing the substance into the flame. When seen through a spectroscope, it shows a large number of lines, of which a green and an orange are most intense and characteristic. Calcium is detected in solution (after removal of the metals by treatment with sulphuretted hydrogen and ammonium sulphide) by boiling with ammonium oxalate and ammonia. The lime is completely thrown down as a white precipitate. Lime is distinguished from the other alkaline earths by forming a sulphate insoluble in dilute alcohol, but completely soluble in a boiling solution of ammonium sulphate.
Lime compounds are for the most part soluble in water or in dilute hydrochloric acid. Calcium fluoride must be first converted into sulphate by evaporation in a platinum dish with sulphuric acid. Insoluble silicates are opened up by fusion with "fusion mixture," as described under Silica.
Separation.—The separation of lime is effected by evaporating with hydrochloric acid, to separate silica; and by treating with sulphuretted hydrogen, to remove the second group of metals. If the substance contains much iron, the solution is next oxidised by boiling with a little nitric acid; and the iron, alumina, &c., are removed as basic acetates. The filtrate is treated with ammonia and sulphuretted hydrogen, and allowed to settle. The filtrate from this is heated to boiling, treated with a solution of ammonium oxalate in excess, boiled for five or ten minutes, allowed to settle for half an hour, and filtered. The precipitate contains all the lime as calcium oxalate.
The precipitate of calcium oxalate is washed with hot water, dried, transferred to a weighed platinum crucible, and ignited at[Pg 322] a temperature not above incipient redness. This ignition converts the oxalate into carbonate, with evolution of carbonic oxide, which burns at the mouth of the crucible with a blue flame.[92] Generally a small quantity of the carbonate is at the same time converted into lime. To reconvert it into carbonate, moisten with a few drops of ammonic carbonate solution, and dry in a water-oven. Heat gently over a Bunsen burner, cool, and weigh. The substance is calcium carbonate (CaCO3), and contains 56 per cent. of lime (CaO). It is a white powder, and should show no alkaline reaction with moistened litmus-paper.
Where the precipitate is small, it is better to ignite strongly over the blowpipe, and weigh directly as lime. With larger quantities, and when many determinations have to be made, it is easier to make the determination volumetrically.
These are carried out either by dissolving the oxalate at once in dilute sulphuric acid, and titrating with permanganate of potassium solution; or by calcining it to a mixture of lime and carbonate, and determining its neutralising power with the standard solutions of acid and alkali.
Titration with Permanganate of Potassium Solution.—This solution is made by dissolving 5.643 grams of the salt in water, and by diluting to 1 litre; 100 c.c. are equivalent to 0.5 gram of lime. The solution is standardised by titrating a quantity of oxalic acid about equivalent to the lime present in the assay; 0.5 gram of lime is equivalent to 1.125 gram of crystallised oxalic acid. The standardising may be done with iron. The standard found for iron multiplied by 0.5 gives that for lime.
The process is as follows:—The calcium oxalate (having been precipitated and washed, as in the gravimetric process) is washed through the funnel into a flask with hot dilute sulphuric acid, boiled till dissolved, diluted to 200 c.c. with water, and heated to about 80° C. The standard solution of "permanganate" is then run in, (not too quickly, and with constant shaking) until a permanent pink tinge is produced. The c.c. used multiplied by the standard, and divided by the weight of the substance taken, will give the percentage of lime.
Estimation of Lime by Alkalimetry.—The methods of determining the amount of an alkali or base by means of a standard acid solution, or, conversely, of determining an acid by means of a standard alkaline solution, are so closely related that[Pg 323] they are best considered under one head. The same standard solution is applicable for many purposes, and, consequently, it is convenient to make it of such strength that one litre of it shall equal an equivalent in grams of any of the substances to be determined. Such solutions are termed normal. For example, a solution of hydrochloric acid (HCl = 36.5) containing 36.5 grams of real acid per litre, would be normal and of equivalent strength to a solution containing either 17 grams of ammonia (NH3 = 17) or 40 grams of sodic hydrate (NaHO = 40) per litre. It will be seen in these cases that the normal solution contains the molecular weight in grams per litre; and, if solutions of these strengths be made, it will be found that they possess equal neutralising value.
If, now, a solution containing 98 grams of sulphuric acid (H2SO4 = 98) per litre be made, it will be found to have twice the strength of the above solution, that is, 100 c.c. of the soda would only require 50 c.c. of the acid to neutralise it. The reason for this will be seen on inspecting the equations:—
NaHO + HCl = NaCl + H2O.
2NaHO + H2SO4 = Na2SO4 + 2H2O.
Acids like sulphuric acid are termed bibasic, and their equivalent is only half the molecular weight. Thus, a normal solution of sulphuric acid would contain 49 grams (98/2) of real acid per litre. Similarly, lime and most of the bases are bibasic, as may be seen from the following equations; hence their equivalent will be half the molecular weight.
2HCl + CaO = CaCl2 + H2O.
2HCl + MgO = MgCl2 + H2O.
The standard normal solution of hydrochloric acid is made by diluting 100 c.c. of the strong acid to one litre with water. This will be approximately normal. In order to determine its exact strength, weigh up 3 grams of recently ignited pure sodium carbonate or of the ignited bicarbonate. Transfer to a flask and dissolve in 200 c.c. of water; when dissolved, cool, tint faintly yellow with a few drops of a solution of methyl orange, and run in the standard "acid " from a burette till the yellow changes to a pink. Read off the number of c.c. used, and calculate to how much sodium carbonate 100 c.c. of the "acid" are equivalent. If the "acid" is strictly normal, this will be 5.3 grams. It will probably be equivalent to more than this. Now calculate how much strictly normal "acid" would be equivalent to the standard[Pg 324] found. For example: suppose the standard found is 5.5 gram of sodium carbonate, then—
5.3 : 5.5 :: 100 : x
(where x is the quantity of normal "acid" required).
x = 103.8 c.c.
To get the "acid" of normal strength, we should then add 3.8 c.c. of water to each 100 c.c. of the standard solution remaining. Suppose there were left 930 c.c. of the approximate "acid," 35.3 c.c. of water must be added and mixed. It should then be checked by another titration with pure sodium carbonate.
The standard solution of semi-normal "alkali." The best alkali for general purposes is ammonia, but, since it is volatile (especially in strong solutions), it is best to make it of half the usual strength, or semi-normal. One litre of this will contain 8.5 grams of ammonia (NH3), and 100 c.c. of it will just neutralise 50 c.c. of the normal "acid." Take 100 c.c. of dilute ammonia and dilute with water to one litre. Run into a flask 50 c.c. of the standard "acid," tint with methyl orange, and run in from a burette the solution of ammonia till neutralised. Less than 100 c.c. will probably be used. Suppose 95 c.c. were required, there should have been 100, hence there is a deficiency of five. Then, for each 95 c.c. of standard "ammonia" left, add 5 c.c. of water, and mix well. 100 c.c. will now be equivalent to 50 c.c. of the "acid."
As an example of the application of this method, we may take the determination of lime in limestone, marble, and similar substances.
Determination of Lime in Limestone.—Weigh up 1 gram of the dried sample, and dissolve in 25 c.c. of normal acid, cool, dilute to 100 c.c., and titrate with the semi-normal solution of alkali (using methyl-orange as an indicator). Divide the c.c. of alkali used by 2, subtract from 25, and multiply by 0.028 to find the weight of lime. This method is not applicable in the presence of other carbonates or oxides, unless the weight of these substances be afterwards determined and due correction be made.
Strontia, the oxide of strontium (SrO), occurs in nature as sulphate, in the mineral celestine (SrSO4), and as carbonate in strontianite (SrCO3). It is found in small quantities in limestones, chalk, &c.
Strontia is used in sugar-refining, and for the preparation of coloured lights.
Detection.—It is detected by the crimson colour which its[Pg 325] compounds (when moistened with hydrochloric acid) impart to the flame. The spectrum shows a large number of lines, of which a red, an orange, and a blue are most characteristic.
It resembles lime in many of its compounds, but is distinguished by the insolubility of its sulphate in a boiling solution of ammonium sulphate, and by the insolubility of its nitrate in alcohol. From baryta, which it also resembles, it is distinguished by not yielding an insoluble chromate in an acetic acid solution, by the solubility of its chloride in alcohol, and by the fact that its sulphate is converted into carbonate on boiling with a solution formed of 3 parts of potassium carbonate and 1 of potassium sulphate.
It is got into solution in the same manner as lime. The sulphate should be fused with "fusion mixture," extracted with water, and thoroughly washed. The residue will contain the strontia as carbonate, which is readily soluble in dilute hydrochloric or nitric acid.
Separation.—It is separated (after removal of the silica and metals, as described under Lime) by adding ammonia and ammonia carbonate, and allowing to stand for some hours in a warm place. In the absence of baryta or lime it is filtered off, and weighed as strontium carbonate, which contains 70.17 per cent. of strontia. It is separated from baryta by dissolving in a little hydrochloric acid, adding ammonia in excess, and then acidifying with acetic acid, and precipitating the baryta with potassium bichromate, as described under Baryta. The strontia is precipitated from the filtrate by boiling for some time with a strong solution of ammonic sulphate and a little ammonia. Fifty parts of ammonic sulphate are required for each part of strontia or lime present. The precipitate is filtered off, and washed first with a solution of ammonic sulphate, and then with alcohol. It is dried, ignited and weighed as strontium sulphate.
The determination of strontia in pure solutions is best made by adding sulphuric acid in excess and alcohol in volume equal to that of the solution. Allow to stand overnight, filter, wash with dilute alcohol, dry, ignite at a red heat, and weigh as sulphate (SrSO4). This contains 56.4 per cent. of strontia (SrO); or 47.7 per cent. of strontium.[Pg 326]
Baryta, oxide of barium (BaO), commonly occurs in combination with sulphuric oxide in the mineral barytes or heavy spar (BaSO4), and in combination with carbon dioxide in witherite (BaCO3). These minerals are not unfrequently found in large quantity (associated with galena and other metallic sulphides) in lodes. Small isolated crystals of these are frequently found in mining districts. Barium is a constituent of certain mineral waters. The minerals are recognised by their high specific gravity and their crystalline form.
Compounds of barium are often used by the assayer, more especially the chloride and hydrate. The salts are, with the exception of the sulphate, generally soluble in water or hydrochloric acid. In such solutions sulphuric acid produces a white precipitate of baric sulphate, which is practically insoluble in all acids.
The dioxide (BaO2) is used for the preparation of oxygen. On strong ignition it gives up oxygen, and is converted into baryta (BaO), which, at a lower temperature, takes up oxygen from the air, re-forming the dioxide.
Detection.—Barium is detected by the green colour its salts, especially the chloride, give to the flame. This, viewed through the spectroscope, shows a complicated spectrum, of which two lines in the green are most easily recognised and characteristic. The salts of barium give no precipitate with sulphuretted hydrogen in either acid or alkaline solution, but with sulphuric acid they at once give a precipitate, which is insoluble in acetate of soda. In solutions rendered faintly acid with acetic acid, they give a yellow precipitate with bichromate of potash. These reactions are characteristic of barium.
Baryta is got into solution in the manner described under Lime; but in the case of the sulphate the substance is fused with three or four times its weight of "fusion mixture." The "melt" is extracted with water, washed, and the residue dissolved in dilute hydrochloric acid.
Separation.—The separation is thus effected:—The solution in hydrochloric acid is evaporated to dryness, re-dissolved in hot dilute hydrochloric acid, and sulphuric acid is added to the solution till no further precipitate is formed. The precipitate is filtered off, and digested with a solution of ammonium acetate or of sodium hyposulphite at 50° or 60° C. to dissolve out any lead sulphate. The residue is filtered off, washed, dried, and ignited. The ignited substance is mixed with four or five times its weight of "fusion mixture," and fused in a platinum-dish over the blowpipe[Pg 327] for a few minutes. When cold, it is extracted with cold water, filtered, and washed. The residue is dissolved in dilute hydrochloric acid, and (if necessary) filtered. The solution contains the barium as baric chloride mixed, perhaps, with salts of strontium or lime. To separate these, ammonia is added till the solution is alkaline, and then acetic acid in slight excess. Chromate of baryta is then thrown down, by the addition of bichromate of potash, as a yellow precipitate. It is allowed to settle, filtered and washed with a solution of acetate or of nitrate of ammonia. It is dried, ignited gently, and weighed. It is BaCrO4, and contains 60.47 per cent. of baryta.
The gravimetric determination of baryta, when lime and strontia are absent, is as follows:—The solution, if it contains much free acid, is nearly neutralised with ammonia, and then diluted to 100 or 200 c.c. It is heated to boiling, and dilute sulphuric acid is added till no further precipitation takes place. The precipitate is allowed to settle for a few minutes, decanted through a filter, and washed with hot water; and, afterwards, dried, transferred to a porcelain crucible, and strongly ignited in the muffle or over the blowpipe for a few minutes. It is then cooled, and weighed as sulphate of baryta (BaSO4). It contains 65.67 per cent. of baryta (BaO).
In determining the baryta in minerals which are soluble in acid, it is precipitated direct from the hydrochloric acid solution (nearly neutralised with ammonia) by means of sulphuric acid. The precipitated baric sulphate is digested with a solution of ammonic acetate; and filtered, washed, ignited, and weighed.
The principle and mode of working of this is the same as that given under the Sulphur Assay; but using a standard solution of sulphuric acid instead of one of barium chloride. The standard solution of sulphuric acid is made to contain 32.02 grams of sulphuric acid (H2SO4), or an equivalent of a soluble alkaline sulphate, per litre. 100 c.c. will be equal to 5 grams of baryta.
Five grams of the substance are taken, and the baryta they contain converted into carbonate (if necessary). The carbonate is dissolved in dilute hydrochloric acid. Ten grams of sodium acetate are added, and the solution, diluted to 500 c.c., is boiled, and titrated in the manner described.[Pg 328]
Lead salts must be absent in the titration, and so must strontia and lime. Ferrous salts should be peroxidised by means of permanganate or chlorate of potash. Other salts do not interfere.
Magnesia, the oxide of magnesium (MgO) occurs in nature in the rare mineral periclase (MgO); and hydrated, as brucite (MgH2O2). As carbonate it occurs in large quantity as magnesite (MgCO3), which is the chief source of magnesia. Mixed with carbonate of lime, it forms magnesian limestone and dolomite. It is present in larger or smaller quantity in most silicates; and the minerals, serpentine, talc, steatite and meerschaum are essentially hydrated silicates of magnesia. Soluble magnesian salts occur in many natural waters; more especially the sulphate and the chloride. Kieserite (MgSO4.H2O) occurs in quantity at Stassfurt, and is used in the manufacture of Epsom salts.
Detection.—Magnesia is best detected in the wet way. Its compounds give no colour to the flame, and the only characteristic dry reaction is its yielding a pink mass when ignited before the blowpipe (after treatment with a solution of cobalt nitrate). In solution, it is recognised by giving no precipitate with ammonia or ammonic carbonate in the presence of ammonic chloride, and by giving a white crystalline precipitate on adding sodium phosphate or arsenate to the ammoniacal solution.
Magnesia differs from the other alkaline earths by the solubility of its sulphate in water.
Magnesia is dissolved by boiling with moderately strong acids; the insoluble compounds are fused with "fusion mixture," and treated as described under Silicates.
Separation.—It is separated by evaporating the acid solution to dryness to render silica insoluble, and by taking up with dilute hydrochloric acid. The solution is freed from the second group of metals by means of sulphuretted hydrogen, and the iron, alumina, &c., are removed with ammonic chloride, ammonia, and ammonic sulphide. The somewhat diluted filtrate is treated, first, with ammonia, and then with carbonate of ammonia in slight excess. It is allowed to stand for an hour in a warm place, and then filtered. The magnesia is precipitated from the filtrate by the addition of an excess of sodium phosphate and ammonia. It is allowed to stand overnight, filtered, and washed with dilute ammonia. The precipitate contains the magnesia as ammonic-magnesic phosphate.
In cases where it is not desirable to introduce sodium salts or phosphoric acid into the assay solution, the following method is[Pg 329] adopted. The solution (freed from the other alkaline earths by ammonium carbonate) is evaporated in a small porcelain dish with nitric acid. The residue (after removing the ammonic salts by ignition) is taken up with a little water and a few crystals of oxalic acid, transferred to a platinum dish, evaporated to dryness, and ignited. The residue is extracted with small quantities of boiling water and filtered off; while the insoluble magnesia is washed. The filtrate contains the alkalies. The residue is ignited, and weighed as magnesia. It is MgO.
The solution containing the magnesia is mixed with chloride of ammonium and ammonia in excess. If a precipitate should form, more ammonic chloride is required. Add sodium phosphate solution in excess, stir and allow to stand overnight. Filter and wash the precipitate with dilute ammonia. Dry, transfer to a platinum or porcelain crucible, and ignite (finally at intense redness); cool, and weigh. The substance is magnesic pyrophosphate (Mg2P2O7), and contains 36.04 per cent. of magnesia.
The magnesia having been precipitated as ammonic-magnesic phosphate, which is the usual separation, its weight can be determined volumetrically by the method of titration described under Phosphates.
The same standard solution of uranium acetate is used. Its standard for magnesia is got by multiplying the standard for phosphoric oxide by 0.5493. For example, if one hundred c.c. are equivalent to 0.5 gram of phosphoric oxide, they will be equivalent to (0.5 × .5493) 0.2746 gram of magnesia. The method of working and the conditions of the titration are the same as for the phosphate titration. The quantity of substance taken for assay must not contain more than 0.1 or 0.2 gram of magnesia. After precipitating as ammonic-magnesic phosphate with sodium phosphate, and well washing with ammonia, it is dissolved in dilute hydrochloric acid, neutralised with ammonia, and sodic acetate and acetic acid are added in the usual quantity. The solution is boiled and titrated.
Silica and Insoluble Silicates.—Take one gram of the dried sample and dissolve it in 10 c.c. of dilute hydrochloric acid; filter; wash, dry, and ignite the residue.[Pg 330]
Organic Matter.—If the residue insoluble in hydrochloric acid shows the presence of organic matter, it must be collected on a weighed filter and dried at 100°. On weighing, it gives the combined weights of organic and insoluble matter. The latter is determined by igniting and weighing again. The organic matter is calculated by difference.
Lime.—Where but little magnesia is present, this is determined by titration with standard acid. Take one gram, and dissolve it in 25 c.c. of normal hydrochloric acid. Tint with methyl-orange and titrate with semi-normal ammonia. Divide the quantity of ammonia used by 2, deduct this from 25, and multiply the remainder by 2.8. This gives the percentage of lime. Where magnesia is present, the same method is adopted, and the magnesia (which is separately determined) is afterwards deducted. The percentage of magnesia found is multiplied by 1.4, and the result is deducted from the apparent percentage of lime got by titrating.
Magnesia.—Dissolve 2 grams of the limestone in hydrochloric acid, and separate the lime with ammonia and ammonium oxalate. The filtrate is treated with sodium phosphate, and the magnesia is weighed as pyrophosphate, or titrated with uranium acetate.
Iron.—Dissolve 2 grams in hydrochloric acid, reduce, and titrate with standard permanganate of potassium solution. This gives the total iron. The ferrous iron is determined by dissolving another 2 grams in hydrochloric acid and at once titrating with the permanganate of potassium solution.
Manganese.—Dissolve 20 grams in hydrochloric acid, nearly neutralise with soda, add sodium acetate, boil, and filter. To the filtrate add bromine; boil, and determine the manganese in the precipitate. See page 300.
Phosphoric Oxide.—This is determined by dissolving the ferric acetate precipitate from the manganese separation in hydrochloric acid, adding ammonia in excess, and passing sulphuretted hydrogen. Filter and add to the filtrate "magnesia mixture." The precipitate is collected, washed with ammonia, ignited, and weighed as pyrophosphate.
The oxides of sodium, potassium, lithium, cæsium, and rubidium and ammonia are grouped under this head. Of these cæsia and rubidia are rare, and lithia comparatively so. They are easily distinguished by their spectra. They are characterised by the solubility of almost all their salts in water, and, consequently, are found in the solutions from which the earths and oxides of the metals have been separated by the usual group re-agents.[Pg 331]
The solution from which the other substances have been separated is evaporated to dryness, and the product ignited to remove the ammonic salts added for the purpose of separation. The residue contains the alkali metals generally, as chlorides or sulphates. Before determining the quantities of the particular alkali metals present, it is best to convert them altogether, either into chloride or sulphate, and to take the weight of the mixed salts. It is generally more convenient to weigh them as chlorides. They are converted into this form, if none of the stronger acids are present, by simply evaporating with an excess of hydrochloric acid. Nitrates are converted into chlorides by this treatment. When sulphates or phosphates are present, the substance is dissolved in a little water, and the sulphuric or phosphoric acid precipitated with a slight excess of acetate of lead in the presence of alcohol. The solution is filtered, and the excess of lead precipitated with sulphuretted hydrogen. The filtrate from this is evaporated to dryness with an excess of hydrochloric acid, and the residue, consisting of the mixed chlorides, is gently ignited and weighed. In many cases (such as the analysis of slags and of some natural silicates where the percentage of alkalies is small) the percentage of soda and potash (which most commonly occur) need not be separately determined. It is sufficient to report the proportion of mixed alkalies; which is thus ascertained:—Dissolve the ignited and weighed chlorides in 100 c.c. of distilled water, and titrate with the standard solution of silver nitrate (using potassic chromate as indicator) in the manner described under Chlorine. The c.c. of silver nitrate used gives the weight in milligrams of the chlorine present. Multiply this by 0.775, and deduct the product from the weight of the mixed chlorides. This will give the combined weight of the alkalies (Na2O and K2O) present. For example, 0.0266 gram of mixed chlorides required on titrating 14.2 c.c. of silver nitrate, which is equivalent to 0.0142 gram of chlorine. This multiplied by 0.775 gives 0.0110 to be deducted from the weight of the mixed chlorides.
| Mixed chlorides | 0.0266 gram |
| Deduction | 0.0110 " |
| ——— | |
| Mixed alkalies | 0.0156 " |
Assuming this to have been got from 1 gram of a rock, it would amount to 1.56 per cent. of "potash and soda."
The relative proportions of the potash and soda can be ascertained from the same determination. Sodium and potassium chlorides have the following composition:[Pg 332]—
| Sodium | 39.38 | Potassium | 52.46 |
| Chlorine | 60.62 | Chlorine | 47.54 |
| ——— | ——— | ||
| 100.00 | 100.00 |
The percentage of chlorine in the mixed chlorides is calculated. It necessarily falls somewhere between 47.5 and 60.6 per cent., and approaches the one or the other of these numbers as the proportion of the sodium or potassium preponderates. Each per cent. of chlorine in excess of 47.5 represents 7.63 per cent. of sodium chloride in the mixed chlorides. The percentage of potash and soda in the substance can be calculated in the usual way. Sodium chloride multiplied by 0.5302 gives its equivalent of soda (Na2O), and potassium chloride multiplied by 0.6317 gives its equivalent of potash (K2O).
The weight of sodium chloride in the mixed chlorides is also calculated thus:—Take the same example for illustration. Multiply the chlorine found by 2.103. This gives—
(0.0142×2.103) = 0.02987.
From the product deduct the weight of the mixed chlorides found—
| Product | 0.02987 |
| Mixed chlorides | 0.02660 |
| ——— | |
| Difference | 0.00327 |
The difference multiplied by 3.6288 gives the weight of sodium chloride in the mixture. In this case it equals 0.0118 gram. The potassium chloride is indicated by the difference between this and the weight of the mixed chlorides. It equals 0.0148 gram. We have now got—
| Sodium chloride | 0.0118 gram |
| Potassium chloride | 0.0148 " |
from 1 gram of the rock taken. Multiplying these by their factors we have (Soda = 0.0118×0.5302; Potash 0.0148×0.6317)—
| Soda | = 0.625 per cent. |
| Potash | = 0.935 " |
Concentration of the Alkalies.—With the exception of magnesia, all the other bases are separated from the alkalies in the ordinary course of work without the addition of any re-agent which cannot be removed by simple evaporation and ignition. Consequently, with substances soluble in acids, successive treatment of the solution with sulphuretted hydrogen, ammonia,[Pg 333] ammonic sulphide, and ammonic carbonate, filtering, where necessary, will yield a filtrate containing the whole of the alkalies with ammonic salts and, perhaps, magnesia.
The filtrate is evaporated in a small porcelain dish, with the addition of nitric acid towards the finish. It is carried to dryness and ignited. The residue is taken up with a little water, treated with a few crystals of oxalic acid, and again evaporated and ignited. The alkaline salts are extracted with water, and filtered from the magnesia into a weighed platinum dish. The solution is then evaporated with an excess of hydrochloric acid, ignited at a low red heat, and weighed. The residue consists of the mixed alkaline chlorides.
For substances (such as most silicates and similar bodies) not completely decomposed by acids, Lawrence Smith's method is generally used. This is as follows:—Take from 0.5 to 1 gram of the finely powdered mineral, and mix, by rubbing in the mortar, with an equal weight of ammonium chloride. Then mix with eight times as much pure calcium carbonate, using a part of it to rinse out the mortar. Transfer to a platinum crucible, and heat gently over a Bunsen burner until the ammonic chloride is decomposed (five or ten minutes). Raise the heat to redness, and continue at this temperature for about three quarters of an hour. The crucible must be kept covered. Cool, and turn out the mass into a 4-inch evaporating dish; wash the crucible and cover with distilled water, and add the washings to the dish; dilute to 60 or 80 c.c., and heat to boiling. Filter and wash. Add to the filtrate about 1.5 gram of ammonium carbonate; evaporate to about 40 c.c., and add a little more ammonic carbonate and some ammonia. Filter into a weighed platinum dish, and evaporate to dryness. Heat gently, to drive off the ammonic chloride, and ignite to a little below redness. Cool and weigh. The residue consists of the mixed alkaline chlorides.
Separation of the Alkali-Metals from each other.—Sodium and lithium are separated from the other alkali-metals by taking advantage of the solubility of their chlorides in the presence of platinic chloride; and from one another by the formation of an almost insoluble lithic phosphate on boiling with a solution of sodium phosphate in a slightly alkaline solution. Cæsium, rubidium, and potassium yield precipitates with platinic chloride, which are somewhat soluble, and must be precipitated from concentrated solutions. Cæsium and rubidium are separated from potassium by fractional precipitation with platinum chloride. Their platino-chlorides, being less soluble than that of potassium, are precipitated first. One hundred parts of boiling water dissolve 5.18 of the potassium platino-chloride, 0.634 of the rubidium[Pg 334] salt, and 0.377 of the corresponding cæsium compound. The separation of lithium, cæsium, and rubidium is seldom called for, owing to their rarity. The details of the separation of potassium from sodium are described under Potassium. Ammonia compounds are sharply marked off from the rest by their volatility, and it is always assumed that they have been removed by ignition; if left in the solution, they would count as potassium compounds. They will be considered under Ammonia.
Sodium is the commonest of the alkali metals. It is found in nature chiefly combined with chlorine as "common salt" (NaCl). This mineral is the source from which the various compounds of sodium in use are prepared. Sodium occurs abundantly as nitrate (NaNO3) in Chili saltpetre, and as silicate in various minerals, such as albite (or soda-felspar).
It occurs as fluoride in cryolite (Na3AlF6), and as carbonate in natron, &c. Sulphates are also found. Sodium is very widely diffused, few substances being free from it.
The detection of sodium is easy and certain, owing to the strong yellow colour its salts impart to the flame; this, when viewed by the spectroscope, shows a single yellow line.[93] The extreme delicacy of this test limits its value, because of the wide diffusion of sodium salts. It is more satisfactory to separate the chloride, which may be recognised by its taste, flame coloration, fusibility, and negative action with reagents. The chloride dissolved in a few drops of water gives with potassium metantimoniate, a white precipitate of the corresponding sodium salt.
Sodium salts are dissolved out from most compounds on treatment with water or dilute acids. Insoluble silicates are decomposed and the alkali rendered soluble by Lawrence Smith's method, which has just been described. The separation of the sodium from the mixed chlorides is effected in the following way:—The chlorides are dissolved in a little water and the potassium separated as platino-chloride. The soluble sodium platino-chloride, with the excess of platinum, is boiled, mixed with sulphuric acid, evaporated to dryness, and ignited. On extracting with water, filtering, evaporating, and igniting, sodium sulphate is left, and is weighed as such.
It is more usual, and quite as satisfactory, to calculate the weight of the sodium chloride by difference from that of the mixed chlorides, by subtracting that of the potassium chloride,[Pg 335] which is separately determined. For example, 1 gram of a rock gave—Mixed chlorides, 0.0266 gram, and 0.0486 gram of potassic platino-chloride. This last is equivalent to 0.0149 gram of potassium chloride.
| Mixed chlorides found | 0.0266 |
| Deduct potassium chloride | 0.0149 |
| ——— | |
| Leaves sodium chloride | 0.0117 |
The weight of sodium chloride found, multiplied by 0.5302, gives the weight of the soda (Na2O).
The solution, which must contain no other metal than sodium, is evaporated in a weighed platinum crucible or dish. Towards the finish an excess, not too great, of sulphuric acid is added, and the evaporation is continued under a loosely fitting cover. The residue is ignited over the blowpipe, a fragment of ammonic carbonate being added towards the end, when fumes of sulphuric acid cease to be evolved. This ensures the removal of the excess of acid. The crucible is cooled in the desiccator, and weighed. The substance is sulphate of soda (Na2SO4), and contains 43.66 per cent. of soda (Na2O), or 32.38 per cent. of sodium (Na).
There are various methods used for the different compounds of sodium. There is no one method of general application. Thus with "common salt" the chlorine is determined volumetrically; and the sodium, after deducting for the other impurities, is estimated by difference.
With sodic carbonate and caustic soda, a given weight of the sample is titrated with standard acid, and the equivalent of soda estimated from the alkalinity of the solution.
With sodium sulphate, a modification of the same method is used. To a solution of 3.55 grams of the salt contained in a half-litre flask, 250 c.c. of a solution of baryta water is added. The volume is made up to 500 c.c. with water. The solution is mixed and filtered. Half of the filtrate is measured off, treated with a current of carbonic acid, and then boiled. It is transferred to a half-litre flask, diluted to the mark, shaken up, and filtered. 250 c.c. of the filtrate, representing a quarter of the sample taken, is then titrated with standard acid. The standard acid is made by diluting 250 c.c. of the normal acid to 1 litre. The c.c. of[Pg 336] acid used multiplied by 2 gives the percentage. A correction must be made to counteract the effect of impurities in the baryta as well as errors inherent in the process. This is small, and its amount is determined by an experiment with 3.55 grams of pure sodium sulphate.
Moisture.—Powder and weigh up 10 grams of the sample into a platinum dish. Dry in a water oven for an hour, and afterwards heat to bare redness over a Bunsen burner. Cool, and weigh. The loss gives the water.
Chlorine.—Weigh up two separate lots of 1 gram each; dissolve in 100 c.c. of water, and determine the chlorine by titrating with the standard silver nitrate solution, using chromate of potash as indicator. See Chlorine.
Insoluble Matter.—Dissolve 10 grams of the salt in water with the help of a little hydrochloric acid. Filter off the sediment, wash, ignite, and weigh. This residue is chiefly sand. Dilute the nitrate to 500 c.c.
Lime.—Take 250 c.c. of the filtrate, render ammoniacal and add ammonium oxalate; wash, dry, and ignite the precipitate. Weigh as lime (CaO).
Magnesia.—To the filtrate from the lime add phosphate of soda. Allow to stand overnight, filter, wash with dilute ammonia, dry, ignite, and weigh as pyrophosphate.
Sulphuric Oxide.—To the remaining 250 c.c. of the filtrate from the "insoluble," add an excess of barium chloride. Collect, wash, dry, ignite, and weigh the barium sulphate.
Sodium.—It is estimated by difference.
The following may be taken as an example:—
| Moisture | 0.35 |
| Insoluble matter | 0.40 |
| Lime | 0.40 |
| Magnesia | 0.05 |
| Sulphuric oxide | 0.60 |
| Chlorine | 59.60 |
| Sodium | 38.60 |
| ——— | |
| 100.00 |
Potassium occurs in nature as chloride, in the mineral sylvine (KCl), and more abundantly combined with magnesium chloride, in earnallite [Pg 337](KCl.MgCl2.6H2O). It occurs as nitrate in nitre (KNO3), and as silicate in many minerals, such as orthoclase (or potash-felspar) and muscovite (or potash-mica).
Potassium compounds are detected by the characteristic violet colour they impart to the flame. The presence of sodium salts masks this tint, but the interference can be neutralised by viewing the flame through a piece of blue glass. Viewed through the spectroscope, it shows a characteristic line in the red and another in the violet. These, however, are not so easy to recognise or obtain as the sodium one. Concentrated solutions of potassium salts give a yellow crystalline precipitate with platinum chloride, and a white crystalline one with the acid tartrate of soda. For these tests the solution is best neutral. These tests are only applicable in the absence of compounds other than those of potassium and sodium.
This process serves for its separation from sodium. Take 1 gram of the sample and dissolve it in an evaporating dish with 50 c.c. of water. Acidify with hydrochloric acid in quantity sufficient (if the metals are present as chlorides) to make it acid, or, if other acids are present, in at least such quantity as will provide the equivalent of chlorine. Add 3 grams of platinum, in solution as platinum chloride, and evaporate on a water-bath to a stiff paste, but not to dryness. Moisten with a few drops of platinic chloride solution without breaking up the paste by stirring. Cover with 20 c.c. of strong alcohol, and wash the crystals as much as possible by rotating the dish. Allow to settle for a few moments, and decant through a filter. Wash in the same way two or three times until the colour of the filtrate shows that the excess of the platinum chloride used is removed. Wash the precipitate on to the filter with a jet of alcohol from the wash-bottle; clean the filter-paper, using as little alcohol as possible. Dry in the water-oven for an hour. Brush the precipitate into a weighed dish, and weigh it. It is potassium platino-chloride (K2PtCl6), and contains 16.03 per cent. of potassium, or 30.56 per cent. of potassium chloride (KCl), which is equivalent to 19.3 per cent. of potash (K2O).
If the filter-paper is not free from precipitate, burn it and weigh separately. The excess of weight over that of the ash will be due to platinum and potassic chloride (Pt and 2KCl). This multiplied by 1.413 will give the weight of the potassic platino-chloride from which it was formed. It must be added to the weight of the main precipitate.
The mixed alkaline chlorides obtained in the usual course of[Pg 338] analysis are treated in this manner; the quantity of platinum added must be about three times as much as the mixed chlorides weigh.
These are the same as with soda.
Examination of Commercial Carbonate of Potash.—The impurities to be determined are moisture, silica, and insoluble matter, chlorine, sulphuric oxide, and oxide of iron. These determinations are made in the ways described under the examination of common salt.
The potassium is determined by converting it into chloride and precipitating with platinum chloride, &c., as just described.
Available Alkali.—Weigh up 23.5 grams of the sample, dissolve in water, and make up to 500 c.c. Take 50 c.c., tint with methyl orange, and titrate with the normal solution of acid. The c.c. of acid used multiplied by 2 gives the percentage of available alkali calculated as potash (K2O).
Soda.—This is calculated indirectly in the following way:—Deduct from the potassium found the quantity required for combination with the chlorine and sulphuric oxide present, and calculate the remainder to potash (K2O). The apparent surplus excess of available alkali is the measure of the soda present.
Carbon Dioxide.—The c.c. of acid used in the available alkali determination, multiplied by 2.2 and divided by 2.35, gives the percentage of carbon dioxide.
Lithia, the oxide of lithium (Li2O), occurs in quantities of 3 or 4 per cent. in various silicates, such as lepidolite (or lithia-mica), spodumene, and petalite. It also occurs as phosphate in triphyline. It is a constituent of the water of certain mineral springs. A spring at Wheal Clifford contained as much as 0.372 gram of lithium chloride per litre. In small quantities, lithia is very widely diffused.
The Detection of lithia is rendered easy by the spectroscope; its spectrum shows a red line lying about midway between the yellow sodium line and the red one of potassium. It also shows a faint yellow line. The colour of the flame (a crimson) is characteristic.
The reactions of the lithium compounds lie between those of the alkalies and of the alkaline earths. Solutions are not precipitated by tartaric acid nor by platinic chloride. The oxide is[Pg 339] slowly soluble in water. The carbonate is not freely soluble. Lithia is completely precipitated by sodic phosphate, especially in hot alkaline solutions.
In its determination the mixed alkaline chlorides obtained in the separation of the alkalies are dissolved in water, a solution of soda is added in slight excess, and the lithia precipitated with sodic phosphate. Before filtering, it is evaporated to dryness and extracted with hot water rendered slightly ammoniacal. The residue is transferred to a filter, dried, ignited, and weighed. The precipitate is lithium phosphate (3Li2O, P2O5), and contains 38.8 per cent. of lithia. The separation of lithia from magnesia is not given by the usual authorities. Wohler recommends evaporating the solution to dryness with carbonate of soda. On extracting the residue with water, the lithia dissolves out and is determined in the filtrate. One hundred parts of water dissolve, at the ordinary temperature, 0.769 parts of lithium carbonate (Li2CO3); the basic magnesia compound is almost insoluble in the absence of carbon dioxide and ammonium salts.
The oxide of caesium, caesia (Cs2O), is found associated with lithia in lepidolite, &c., and, together with rubidium, in many mineral waters. The mineral pollux is essentially a silicate of alumina and caesia; it contains 34.0 per cent. of the latter oxide.
Caesium is best detected by the spectroscope, its spectrum being characterised by two lines in the blue and one in the red; the latter is about midway between the lithium and sodium lines.
If not detected by the spectroscope, or specially looked for, caesia would, in the ordinary course of work, be separated with the potash and weighed as potassium platino-chloride.
Caesia is separated from all the other alkalies by adding to the acid solution of the mixed chlorides a strongly acid cold solution of antimonious chloride. The acid used must be hydrochloric. The caesium is precipitated as a white crystalline precipitate (CsCl.SbCl3), which is filtered off, and washed, when cold, with strong hydrochloric acid; since it is decomposed by water or on warming. The precipitate is washed into a beaker, and treated with sulphuretted hydrogen; after filtering off the sulphide of antimony, the solution leaves, on evaporation, the caesium as chloride.[Pg 340]
Rubidium occurs widely diffused in nature, but in very small quantities. It is generally associated with caesium.
It is detected by the spectroscope, which shows two violet lines and two dark red ones. Like caesium, it is precipitated with platinic chloride, and in the ordinary course of work would be weighed as potassium. It is separated from potassium by fractional precipitation with platinic chloride. Rubidium platino-chloride is much less soluble than the potassium salt.
It is usual to look upon the salts of ammonia as containing a compound radical (NH4 = Am), which resembles in many respects the metals of the alkalies. Ammonium occurs in nature as chloride in sal ammoniac (AmCl), as sulphate in mascagnine (Am2SO4), as phosphate in struvite (AmMgPO4.12H2O). Minerals containing ammonium are rare, and are chiefly found either in volcanic districts or associated with guano. Ammonia and ammonium sulphide occur in the waters of certain Tuscan lagoons, which are largely worked for the boracic acid they contain. The crude boracic acid from this source contains from 5 to 10 per cent. of ammonium salts. It is from these that the purer forms of ammonium compounds of commerce known as "from volcanic ammonia" are derived. But the bulk of the ammonia of commerce is prepared from the ammoniacal liquors obtained as bye-products in the working of certain forms of blast furnaces and coke ovens, and more especially in gas-making.
Ammonia hardly comes within the objects of assaying; but it is largely used in the laboratory, and the assayer is not unfrequently called on to determine it. Ammonium salts are mostly soluble in water. In strong solutions they give a yellow precipitate of ammonium platino-chloride on the addition of chloride of platinum; and with the acid tartrate of soda yield a white precipitate of hydric ammonic tartrate. These reactions are similar to those produced with potassium compounds.
Heated with a base, such as lime or sodic hydrate, ammonium salts are decomposed, yielding ammonia gas (NH3), which is readily soluble in water. The solution of this substance is known as ammonic hydrate or "ammonia."
They are volatilised on ignition; either with, or without, decomposition according to the acid present. This fact is of importance in analytical work; since it allows of the use of alkaline[Pg 341] solutions and reagents which leave nothing behind on heating. It must be remembered, however, that, although ammonic chloride is volatile, it cannot be volatilised in the presence of substances which form volatile chlorides without loss of the latter. For example: ferric oxide and alumina are thus lost, volatilising as chlorides; and there are some other compounds (notably ammonic magnesic arsenate) which on heating to redness suffer reduction. The presence of ammonic chloride in such cases must be avoided.
Detection.—Compounds of ammonium are detected by their evolving ammonia when mixed or heated with any of the stronger bases. The ammonia is recognised by its odour, by its alkaline reaction with litmus paper, and by yielding white fumes, when brought in contact with fuming acid. In consequence of the use of ammonium salts and ammonia as reagents, it is necessary to make a special test for and determination of ammonium.[94] In the ordinary course of work it will be "lost on ignition." The determination presents little difficulty, and is based on the method used for its detection.
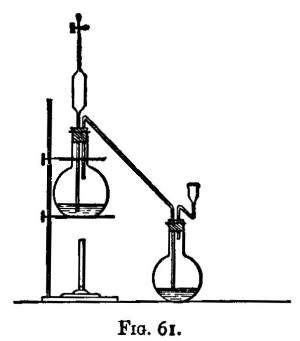
Solution and Separation.—Although ammonium salts are soluble in water, there is no necessity for dissolving them. The compound containing the ammonia is boiled with an alkaline solution; and the liberated ammonia condensed and collected. The substance is weighed out into a flask of about 200 c.c. capacity. The flask is closed with a rubber cork perforated to carry a 20 c.c. pipette and a bulb exit tube. The latter is connected with a receiver, which is a small flask containing dilute hydrochloric acid (fig. 61). The flask containing the substance is corked, and the greater part of the soda solution is run in from the pipette. The solution is then boiled. The ammonia volatilises, and is carried over into the hydrochloric acid, with which it combines to form ammonic chloride. The distillation is carried on gently until the bulk of the liquid is driven over. The ammonia in the receiver will be mixed only with the excess of hydrochloric acid. This separation is used in all determinations.[Pg 342]
The contents of the flask are transferred to a weighed platinum dish, and evaporated on the water-bath. It is dried until the weight is constant. The chloride of ammonium remains as a white mass which, after cooling in a desiccator, is weighed. It contains 33.72 per cent. of ammonium (NH4), or 31.85 per cent. of ammonia (NH3). On heating over the Bunsen burner it is completely volatilised, leaving no residue.
Weigh up 1.7 gram of the substance and place it in the flask. Measure off 50 c.c. of the normal solution of acid, place them in the receiver, and dilute with an equal volume of water. Run in through the pipette (by opening the clip) 20 c.c. of a strong solution of soda, boil until the ammonia has passed over, and then aspirate a current of air through the apparatus. Disconnect the receiver, and tint its contents with methyl orange. Titrate the residual acid with a semi-normal solution of alkali. Divide the c.c. of the "alkali" solution used by 2, and deduct from the 50 c.c. The difference will give the number of c.c. of the normal acid solution neutralised by the ammonia distilled over. Each c.c. of "acid" so neutralised, represents 1 per cent. of ammonia in the sample. If the results are to be reported as ammonium, 1.8 gram of the sample is taken instead of 1.7 gram.
This is effected by means of "Nessler's" reagent, which strikes a brown colour with traces of ammonia, even with a few hundredths of a milligram in 100 c.c. of liquid. With larger quantities of ammonia the reagent gives a precipitate. This reagent is a strongly alkaline solution of potassic mercuric iodide; and is thus made:—
Nessler's solution: Dissolve 17 grams of mercuric chloride in 300 c.c. of water; and add the solution to one of 35 grams of potassium iodide in 100 c.c. of water until a permanent precipitate is produced. Both solutions must be cold. Then make up to a litre by adding a 20 per cent. solution of potash. Add more of the mercuric chloride (a little at a time) until a permanent precipitate is again formed. Allow to settle, decant, and use the clear liquor. Four or five c.c. are used for each 100 c.c. of liquid to be tested.
A Standard Solution of Ammonia is made by dissolving 0.315[Pg 343] gram of ammonic chloride in water, and diluting to 100 c.c. Ten c.c. of this are taken and diluted to 1 litre. One c.c. contains 0.01 milligram of ammonia (NH3).
In working, the solution containing the ammonia is diluted to a definite volume, and to such an extent that 50 c.c. of it shall not contain more than 0.02 or 0.03 milligram of ammonia. Fifty c.c. of it are transferred to a Nessler glass and mixed with 2 c.c. of Nessler's reagent. The colour is noted, and an estimate made as to the amount of ammonia it indicates. A measured quantity of the standard ammonia, judged to contain about as much ammonia as that in the assay, is then put into another Nessler glass. It is diluted to 50 c.c. with water, and mixed with 2 c.c. of "Nessler." After standing a minute or two, the colours in the two glasses are compared. If the tints are equal, the assay is finished; but if the standard is weaker or stronger than the assay, another standard, containing more or less ammonia, as the case may be, must be prepared and compared with the assay. Two such experiments will generally be sufficient; but, if not, a third must be made. The addition of more standard ammonia to the solution to which the "Nessler" has already been added does not give a satisfactory result.
When the ammonia in 50 c.c. has been determined, that in the whole solution is ascertained by a suitable multiplication. By 10, for example, if the bulk was 500 c.c., or by 20 if it was a litre.
Distilled water is used throughout. It must be free from ammonia; and is best prepared by distilling an ammonia-free spring water.
[90] Al2Cl6 + 3Na2S2O3 + 3H2O = Al2(HO)6 + 6NaCl + 3S + 3SO2
[91] 3BeO,Al2O3,6SiO2
[92] CaC2O4 = CaCO3+CO.
[93] Resolved into two with a powerful spectroscope.
[94] Ammonium compounds are frequently produced when dissolving metals in nitric acid; or when nitrates are heated in the presence of the metals.
Oxygen occurs in nature in the free state, forming 23 per cent. by weight, or 21 per cent. by volume of the atmosphere; but, since it is a gas, its presence is easily overlooked and its importance underestimated. Except in the examination of furnace-gases, &c., the assayer is not often called upon to determine its quantity, but it forms one of his most useful reagents, and there are many cases where he cannot afford to disregard its presence. It occurs not only in the air, but also dissolved in water; ordinary waters containing on an average 0.00085 per cent. by weight, or 0.85 parts per 100,000.
Chemically, it is characterised by its power of combining, especially at high temperatures, with the other elements, forming an important class of compounds called oxides. This combination, when rapid, is accompanied by the evolution of light and heat; hence oxygen is generally called the supporter of combustion. This property is taken advantage of in the operation of calcining, scorifying, cupelling, &c. The importance of a free access of air in all such work is seen when it is remembered that 1 litre of air contains 0.2975 gram of oxygen, and this quantity will only oxidise 0.1115 gram of carbon, 0.2975 gram of sulphur, or 3.849 grams of lead.
Oxidation takes place at the ordinary temperature with many substances. Examples of such action are seen in the weathering of pyrites, rusting of iron, and (in the assay office) the weakening of solutions of many reducing agents.
For methods of determining the percentage of oxygen in gases, for technical purposes, the student is referred to Winkler & Lunge's "Technical Gas Analysis."[Pg 345]
Oxides are abundant in nature, almost all the commonly occurring bodies being oxidised. Water (H2O) contains 88.8 per cent. of oxygen; silica, lime, alumina, magnesia, and the other earths are oxides, and the oxides of the heavier metals are in many cases important ores; as, for example, cassiterite (SnO2), hæmatite (Fe2O3), magnetite (Fe3O4), and pyrolusite (MnO2). In fact, the last-named mineral owes its value to the excess of oxygen it contains, and may be regarded as an ore of oxygen rather than of manganese.
Most of the metals, when heated to redness in contact with air, lose their metallic lustre and become coated with, or (if the heating be prolonged) altogether converted into, oxide. This oxide was formerly termed a "calx," and has long been known to weigh more than the metal from which it was obtained. For example, one part by weight of tin becomes, on calcining, 1.271 parts of oxide (putty powder). The student will do well to try the following experiments:—Take 20 grams of tin and heat them in a muffle on a scorifier, scraping back the dross as it forms, and continuing the operation until the whole of the metal is burnt to a white powder and ceases to increase in weight.[95] Take care to avoid loss, and, when cold, weigh the oxide formed. The oxide should weigh 25.42 grams, which increase in weight is due to the oxygen absorbed from the air and combined with the metal. It can be calculated from this experiment (if there has been no loss) that oxide of tin contains 21.33 per cent. of oxygen and 78.67 per cent. of tin. Oxidation is performed with greater convenience by wet methods, using reagents, such as nitric acid, which contain a large proportion of oxygen loosely held. Such reagents are termed oxidising agents. Besides nitric acid, permanganate of potash, bichromate of potash, and peroxide of hydrogen are largely used for this purpose. One c.c. of nitric acid contains as much oxygen as 2.56 litres of air, and the greater part of this is available for oxidising purposes. Try the following experiment:—Take 2 grams of tin and cover in a weighed Berlin dish with 20 c.c. of dilute nitric acid, heat till decomposed, evaporate to dryness, ignite, and weigh. The 2 grams of tin should yield 2.542 grams of oxide. The increase in weight will be proportionally the same as in the previous experiment by calcination, and is due to oxygen, which in this case has been derived from the nitric acid.[Pg 346]
The percentage of oxygen in this oxide of tin (or in any of the oxides of the heavier metals) may be directly determined by heating such oxides in a current of hydrogen, and collecting and weighing the water formed.
It is found by experiment that 88.86 parts by weight of oxygen, combining with 11.14 parts of hydrogen, form 100 parts of water; so that from the weight of water formed it is easy to calculate the amount of oxygen the oxide contained.
Take 1 gram of the dried and powdered oxide and place it in a warm dry combustion tube. Place the tube in a furnace, and connect at one end with a hydrogen apparatus provided with a sulphuric acid bulb for drying the gas, and at the other with a weighed sulphuric acid tube for collecting the water formed. The apparatus required is shown in fig. 62. Pass hydrogen through the apparatus, and, when the air has been cleared out, light the furnace. Continue the heat and current of hydrogen for half an hour (or longer, if necessary). Allow to cool. Draw a current of dry air through the weighed tube. Weigh. The increase in weight gives the amount of water formed, and this, multiplied by 0.8886, gives the weight of the oxygen. The percentage of oxygen thus determined should be compared with that got by the oxidation of the metal. It will be practically the same. The following results can be taken as examples:—
Twenty grams of tin, calcined as described, gave 25.37 grams of oxide.
Two grams of tin, oxidised with nitric acid and ignited, gave 2.551 grams of oxide.
One gram of the oxide of tin, on reduction in a current of hydrogen, gave 0.2360 gram of water (equivalent to 0.2098 gram of oxygen), and left 0.7900 gram of metal.
Ten grams of ferrous sulphate gave, on strong ignition, 2.898 grams of ferric oxide (Fe2O3)[96] instead of 2.877.
The student should similarly determine the percentage of oxygen in oxides of copper and iron. The former oxide may be prepared by dissolving 5 grams of copper in 50 c.c. of dilute nitric[Pg 347] acid, evaporating to dryness, and strongly igniting the residue. The oxide of iron may be made by weighing up 10 grams of powdered ferrous sulphate (= to 2.014 grams of iron) and heating, at first gently, to drive off the water, and then at a red heat, until completely decomposed. The weight of oxide, in each case, should be determined; and the percentage of oxygen calculated. Compare the figures arrived at with those calculated from the formula of the oxides, CuO and Fe2O3.
It would be found in a more extended series of experiments that the same metal will, under certain conditions, form two or more oxides differing among themselves in the amount of oxygen they contain. These oxides are distinguished from one another by such names as "higher" and "lower oxides," "peroxides," "protoxides," "dioxides," &c.
The oxides may be conveniently classified under three heads:—
(1) Those that are reduced to metal by heat alone, such as the oxides of mercury, silver, platinum, gold, &c.;
(2) Those which are reduced by hydrogen at a red heat, which includes the oxides of the heavy metals;
(3) Those which are not reduced by these means, good examples of which are silica, alumina, the alkalies, and the alkaline earths.
Another important classification is into acid, basic and neutral oxides. The oxides of the non-metallic elements, such as sulphur, carbon, phosphorus, &c., are, as a rule, acid; and the more oxygen they contain, the more distinctly acid they are. The oxides of the metals are nearly all basic; and, as a rule, the less oxygen they contain, the more distinctly basic they are.
The basic oxides, which are soluble in acids, give rise to the formation of salts when dissolved therein. During the solution, water is formed, but no gas is evolved. The oxide dissolved in each case neutralizes an equivalent of the acid used for solution.[97] The basic properties of many of these can be taken advantage of for their determination. This is done in the case of soda, potash, lime, &c., by finding the quantity of acid required to neutralize a given weight of the substance.
There are some oxides which, under certain conditions, are acid to one substance (a stronger base) and basic to another (a stronger acid). For example, the oxides of lead and of tin, as also alumina, dissolve in caustic soda, acting as acids; whilst, on[Pg 348] the other hand, they combine with sulphuric or hydrochloric acid, playing the part of bases.
The oxides known as "earths," when ignited, are many of them insoluble in acids, although easily dissolved before ignition.
It is common in complete analyses of minerals to meet with cases in which the sum total of the elements found falls short of the amount of ore taken; and here oxygen must be looked for. For example, this occurs in the case of a mixture of pyrites with oxide of iron, or in a mixture of sulphides and sulphates. The state in which the elements are present, and the percentage (say of sulphides and sulphates) can in many cases be determined; but this is not always required. When the difference between the sum total and the elements found is small, it is reported as "oxygen and loss." When, however, it is considerable, the oxygen may be reported as such; and its amount be either determined directly in the way already described, or calculated from the best determination that can be made of the relative amounts of oxides, sulphides, sulphates, &c., present. Such cases require a careful qualitative analysis to find out that the substance is present; and then the separation of each constituent is made as strictly as possible. These remarks apply especially to ores of the heavy metals. The separation of the constituents is effected with suitable solvents applied in proper order. The soluble sulphates, for example, are extracted with water; the oxides by the dilute acids or alkalies in which they are known to be soluble. The oxygen in the sulphates and oxides thus obtained is estimated by determining the sulphur and metals in the solutions, and calculating the amount of oxygen with which they combine. The metals of the earths and alkalies are almost invariably present as oxides, and are reported as such; except it is known that they are present in some other form, such as fluoride or chloride. Thus, silica, alumina, lime, water, &c., appear in an analysis; even in those cases where "oxygen and loss" is also mentioned. As an example of such a report, take the following analysis of Spanish pyrites:—
| Sulphur | 49.00 |
| Iron | 43.55 |
| Copper | 3.20 |
| Arsenic | 0.47 |
| Lead | 0.93 |
| Zinc | 0.35 |
| Lime | 0.10 |
| Silica, &c. | 0.63 |
| Water | 0.70 |
| Oxygen and loss | 1.07 |
| ——— | |
| 100.00 |
The following example will illustrate the mode of calculating and reporting. A mineral, occurring as blue crystals soluble in water, and found on testing to be a mixed sulphate of iron and copper, gave on analysis the following results:—
| Water | 44.51 | per cent. |
| Sulphuric oxide | 28.82 | " |
| Copper | 8.44 | " |
| Ferrous iron | 11.81 | " |
| Ferric iron | 0.38 | " |
| Zinc | 0.28 | " |
| ——— | ||
| 94.24 |
There is here a deficiency of 5.76 per cent. due to oxygen. Nothing else could be found, and it is known that in the sulphates the metals exist as oxides. By multiplying the weight of the copper by 1.252, the weight of copper oxide (CuO) will be ascertained; in this case it equals 10.57 per cent. The ferrous iron multiplied by 1.286 will give the ferrous oxide (FeO); in this case 15.19 per cent. The ferric iron multiplied by 1.428 will give the ferric oxide (Fe2O3); in this case 0.54 per cent. The zinc multiplied by 1.246 will give the zinc oxide (ZnO); in this case it equals 0.35 per cent. The analysis will be reported as—
| Water | 44.51 | |
| Sulphuric oxide | 28.82 | |
| Copper oxide | 10.57 | equal to copper 8.44% |
| Ferrous oxide | 15.19 | |
| Ferric oxide | 0.54 | |
| Zinc oxide | 0.35 | |
| ——— | ||
| 99.98 |
The following (A) is an analysis of a sample of South American copper ore, which will serve as a further illustration. The analysis showed the presence of 6.89 per cent. of ferrous oxide, and some oxide of copper.
The analysis (B) is that of an ore from the same mine after an imperfect roasting. It will be seen that the carbonates have been converted into sulphates. If the total sulphur simply had been determined, and the sulphate overlooked, the "oxygen and loss" would have been 5.65 per cent., an amount which would obviously require an explanation.[Pg 350]
| A. | B. | ||||
| Water | 0.25 | 0.59 | |||
| Organic matter | 0.54 | — | |||
| Sulphur | 29.50 | 21.33 | |||
| Copper | 10.92 | 9.80 | {Copper | 9.57 | |
| {Copper oxide | 0.28 | ||||
| Iron | 32.09 | 39.73 | {Iron | 34.32 | |
| {Ferric oxide | 7.73 | ||||
| Lead | 0.35 | 0.12 | |||
| Zinc | 0.86 | 0.69 | |||
| Cobalt | 0.06 | 0.11 | |||
| Lime | 5.25 | 7.69 | |||
| Magnesia | 2.33 | 2.55 | |||
| Sulphuric oxide | 1.00 | 5.30 | |||
| Carbon dioxide | 8.87 | — | |||
| "Insoluble silicates" | 5.12 | 8.38 | |||
| Oxygen and loss | 2.86 | 2.47 | |||
| ——— | Potash | 0.15 | |||
| 100.00 | Soda | 1.09 | |||
| ——— | |||||
| 100.00 |
Water occurs in minerals in two forms, free and combined. The term "moisture" ought, strictly, to be limited to the first, although, as has already been explained, it is more convenient in assaying to apply the term to all water which is driven off on drying at 100° C. The combined water is really a part of the mineral itself, although it may be driven off at a high temperature, which varies with the base. In some cases a prolonged red heat is required; whilst with crystallised salts it is sometimes given off at the ordinary temperatures. This latter phenomenon, known as efflorescence, is mostly confined to artificial salts.
The determination of the combined water may often be made by simply igniting the substance from which the moisture has been removed. The quantity of water may be determined, either indirectly by the loss, or directly by collecting it in a calcium chloride tube, and weighing. In some cases, in which the loss on ignition does not give simply the proportion of combined water, it can be seen from the analysis to what else the loss is due; and, after a proper deduction, the amount of water can be estimated. For example, 1 gram of crystallised iron sulphate was found to contain on analysis 0.2877 gram of sulphuric oxide; and on igniting another gram, 0.2877 gram of ferric oxide was left. As the salt is known to be made up of ferrous oxide, sulphuric oxide, and combined water, the combined water can be thus calculated: 0.2877 gram of ferric oxide is equal to 0.2589 gram of ferrous[Pg 351] oxide,[98] and consequently, the loss on ignition has been diminished by 0.0288 gram, which is the weight of oxygen absorbed by the ferrous oxide during calcining. The loss on ignition was 0.7123 gram, to which must be added 0.0288 gram; hence 0.7411 gram is the weight of the combined sulphuric oxide and water present. Deducting the weight of sulphuric oxide found, 0.2877 gram, there is left for combined water 0.4534 gram. The composition of 1 gram of the dry salt is then:—
| Water | 0.4534 |
| Sulphuric oxide | 0.2877 |
| Ferrous oxide | 0.2589 |
| ——— | |
| 1.0000 |
The following is another example:—A sample of malachite lost on ignition 28.47 per cent., leaving a residue which was found on analysis to be made up of oxide of copper (equal to 70.16 per cent. on the mineral), and silica and oxide of iron (equal to 1.37 per cent.). Carbon dioxide and water (but nothing else) was found to be present, and the carbon dioxide amounted to 19.64 per cent.; deducting this from the loss on ignition, we have 8.82 as the percentage of water present. The analysis was then reported as follows:—
| Cupric oxide | 70.16 | equal to 56.0% copper. |
| Silica and ferric oxide | 1.37 | |
| Carbon dioxide | 19.64 | |
| Water | 8.82 | |
| ——— | ||
| 99.99 |
Direct Determination of Combined Water.—Transfer about 3 grams of the substance to a piece of combustion tube (8 or 10 inches long), attached (as in fig. 63) at one end to a U-tube containing sulphuric acid, and at the other end to a calcium[Pg 352] chloride tube. The last is weighed previous to the determination. The tube should be warmed to ensure complete dryness, and must be free from a misty appearance. Aspirate a current of air through the apparatus, heat the mineral by means of a Bunsen burner, cautiously at first, and afterwards to redness (if necessary). The water is driven off and condenses in the calcium chloride tube, which is afterwards cooled and weighed. The increase in weight is due to the water. If the substance gives off acid products on heating, it is previously mixed with some dry oxide of lead or pure calcined magnesia.
The assayer is occasionally called on to test water for the purpose of ascertaining the nature and quantity of the salts contained in it, and whether it is or is not fit for technical and drinking purposes.
In mineral districts the water is generally of exceptional character, being more or less charged, not only with earthy salts, but also frequently with those of the metals. Distilled water is only used by assayers in certain exceptional cases, so that by many it would be classed among the rarer oxides. Water of ordinary purity will do for most purposes, but the nature and quantity of the impurities must be known.
The following determinations are of chief importance:—
Total Solids at 100° C.—Where simply the amount is required, take 100 c.c. and evaporate on the water-bath in a weighed dish; then dry in the water-oven, and weigh.
Total Solids Ignited.—The above residue is very gently ignited (keeping the heat well below redness), and again weighed. A larger loss than 4 or 5 parts per 100,000 on the water requires an explanation.
Chlorine.—Take 100 c.c. of the water in a porcelain dish, add 2 c.c. of a 5 per cent. solution of neutral potassic chromate, and titrate with a neutral standard solution of nitrate of silver, made by dissolving 4.789 grams of crystallised silver nitrate in distilled water, and diluting to 1 litre. The addition of the nitrate of silver is continued until the yellow of the solution assumes a reddish tint. The reaction is very sharp. Each c.c. of nitrate of silver used is equal to 1 part by weight of chlorine in 100,000 of water. At inland places this rarely amounts to more than 1 in 100,000; but near the sea it may amount to 3 or 5. More than this requires explanation, and generally indicates sewage pollution.
Nitric Pentoxide (N2O5).—It is more generally reported under the heading, "nitrogen as nitrates." Take 250 c.c. of the[Pg 353] water and evaporate to 2 or 3 c.c.; acidulate with a few drops of dilute sulphuric acid, and transfer to a nitrometer (using strong sulphuric acid to wash in the last traces). The sulphuric acid must be added to at least twice the bulk of the liquid. Shake up with mercury. The mercury rapidly flours, and nitric oxide is given off (if any nitrate is present). The volume of the nitric oxide (corrected to normal temperature and pressure), multiplied by 0.25, gives the parts of nitrogen per 100,000; or, multiplied by 0.965, will give the nitric pentoxide in parts per 100,000. In well and spring waters the nitrogen may amount to 0.3 or 0.4 parts per 100,000; or in richly cultivated districts 0.7 or 0.8 parts per 100,000. An excess of nitrates is a suspicious feature, and is generally due to previous contamination.
Ammonia.—Take 500 c.c. of the water and place them in a retort connected with a Liebig's condenser. Add a drop or two of a solution of carbonate of soda and distil over 100 c.c.; collect another 50 c.c. separately. Determine the ammonia in the distillate colorimetrically (with Nessler's solution, as described under Ammonia) and compare with a standard solution of ammonic chloride containing 0.0315 gram of ammonic chloride in 1 litre of water. One c.c. contains 0.01 milligram of ammonia. The second distillate will show little, if any, ammonia in ordinary cases. The amounts found in both distillates are added together, and expressed in parts per 100,000.
Waters (other than rain and tank waters) which contain more than 0.003 per 100,000 are suspicious.
Organic Matter.—The organic matter cannot be determined directly; but for ordinary purposes it may be measured by the amount of permanganate of potassium which it reduces, or by the amount of ammonia which it evolves on boiling with an alkaline permanganate of potassium solution.
A. Albuminoid Ammonia.—To the residue left after distilling the ammonia add 50 c.c. of a solution made by dissolving 200 grams of potash and 8 grams of potassium permanganate in 1100 c.c. of water, and rapidly boiling till the volume is reduced to 1 litre (this should be kept in a well stoppered bottle, and be occasionally tested to see that it is free from ammonia). Continue the distillation, collecting 50 c.c. at a time, until the distillate is free from ammonia. Three or four fractions are generally sufficient. Determine the ammonia colorimetrically as before. If the total albuminoid ammonia does not exceed 0.005 in 100,000, the water may be regarded as clean as regards organic matter; if it amounts to more than 0.015, it is dirty.
B. Oxygen Consumed.—A standard solution of permanganate of potash is made by dissolving 0.395 gram of the salt in water[Pg 354] and diluting to 1 litre. Each c.c. equals 0.1 milligram of available oxygen. The following are also required:—1. A solution of sodium hyposulphite containing 1 gram of the salt (Na2S2O3.5H2O) in 1 litre of water. 2. Dilute sulphuric acid, made by adding one part of the acid to three of water, and titrating with the permanganate solution till a faint pink persists after warming for several hours. 3. Starch paste. 4. Potassium iodide solution.
Take 250 c.c. of the water in a stoppered bottle, add 10 c.c. of sulphuric acid and 10 c.c. of the permanganate, and allow to stand in a warm place for four hours. Then add a few drops of the solution of potassium iodide, and titrate the liberated iodine with "hypo," using starch paste towards the end as an indicator. To standardise the hyposulphite, take 250 c.c. of water and 10 c.c. of sulphuric acid, and a few drops of potassium iodide; then run in 10 c.c. of the "permanganate" solution, and again titrate; about 30 c.c. of the "hypo" will be used. The difference in the two titrations, divided by the last and multiplied by 10, will give the c.c. of permanganate solution used in oxidising the organic matter in the 250 c.c. of water. Each c.c. represents 0.04 parts of oxygen in 100,000.
Metals.—These may for the most part be estimated colorimetrically.
Lead.—Take 100 c.c. of the water in a Nessler tube, and add 10 c.c. of sulphuretted hydrogen water, and compare the tint, if any, against a standard lead solution, as described under Colorimetric Lead. Report in parts per 100,000.
Copper.—Proceed as with the last-mentioned metal; but, if lead is also present, boil down 500 c.c. to about 50 c.c., then add ammonia, filter, and estimate the copper in the blue solution, as described under Colorimetric Copper.
Iron.—Take 50 c.c., or a smaller quantity (if necessary), dilute up to the mark with distilled water, and determine with potassium sulphocyanate, as described under Colorimetric Iron.
Zinc.—Zinc is the only other metal likely to be present; and, since it cannot be determined colorimetrically, it must be separately estimated during the examination of the "total solids."
Examination of "Total Solids."—Evaporate 500 c.c. to dryness with a drop or two of hydrochloric acid. Take up with hydrochloric acid, filter, ignite, and weigh the residue as "silica." To the filtrate add a little ammonic chloride and ammonia, boil and filter, ignite, and weigh the precipitate as "oxide of iron and alumina." Collect the filtrate in a small flask, add a few drops of ammonium sulphide or pass sulphuretted hydrogen, cork the flask, and allow to stand overnight; filter, wash, and determine the zinc gravimetrically as oxide of zinc. If copper or lead were[Pg 355] present, they should have been previously removed with sulphuretted hydrogen in the acid solution. To the filtrate add ammonic oxalate and ammonia, boil for some time, allow to stand, filter, wash, ignite, and weigh as "lime." Evaporate the filtrate with nitric acid, and ignite. Take up with a few drops of dilute hydrochloric acid, add baric hydrate in excess, evaporate, and extract with water. The residue contains the magnesia; boil with dilute sulphuric acid, filter, precipitate it with phosphate of soda and ammonia, and weigh as pyrophosphate. The aqueous extract contains the alkalies with the excess of barium. Add sulphuric acid in slight excess, filter, evaporate, and ignite strongly. The residue consists of the sulphates of the alkalies (which are separately determined, as described under Potash).
Sulphuric Oxide (SO3).—Take 200 c.c. and boil to a small bulk with a little hydrochloric acid, filter (if necessary), add baric chloride solution in slight excess to the hot solution, filter, ignite, and weigh as baric sulphate.
Carbon Dioxide (free).—Carbon dioxide exists in waters in two forms, free and combined. The latter generally occurs as bicarbonate, although on analysis it is more convenient to consider it as carbonate, and to count the excess of carbon dioxide with the free. The method is as follows:—To determine the free carbon dioxide, take 100 c.c. of the water, place them in a flask with 3 c.c. of a strong solution of calcium chloride and 2 c.c. of a solution of ammonic chloride, next add 50 c.c. of lime-water. The strength of the lime-water must be known. Make up to 200 c.c. with distilled water, stop the flask, and allow the precipitate to settle. Take out 100 c.c. of the clear solution with a pipette, and titrate with the standard solution of acid.[99] The number of c.c. required, multiplied by two, and deducted from that required for the 50 c.c. of lime-water, and then multiplied by 0.0045, will give the carbon dioxide present other than as normal carbonates.
Carbon Dioxide combined as normal carbonate.—100 c.c. of the water are tinted with phenacetolin or lacmoid; then heated to near boiling, and titrated with standard acid. The number of c.c. used, multiplied by 0.0045, will give the weight in grams of the combined carbon dioxide.
Free Acid.—In some waters (especially those from mining districts) there will be no carbonates. On the contrary, there may be free mineral acid or acid salts. In these cases it is necessary to determine the amount of acid (other than carbon dioxide) present in excess of that required to form normal salts. This is done in the following way:—Make an ammoniacal copper[Pg 356] solution by taking 13 grams of copper sulphate (CuSO4.5H2O), dissolving in water, adding solution of ammonia until the precipitate first formed has nearly dissolved, and diluting to 1 litre. Allow to settle, and decant off the clear liquid. The strength of this solution is determined by titrating against 10 or 20 c.c. of the standard solution of sulphuric acid (100 c.c. = 1 gram H2SO4). The finishing point is reached as soon as the solution becomes turbid from precipitated cupric hydrate. At first, as each drop falls into the acid solution, the ammonia and cupric hydrate combine with the free acid to form ammonic and cupric sulphates; but as soon as the free acid is used up, the ammonia in the next drop not only precipitates an equivalent of cupric hydrate from the solution, but also throws down that carried by itself. This method is applicable in the presence of metallic sulphates other than ferric. The standardising and titration should be made under the same conditions. Since sulphuric acid and sulphates are predominant in waters of this kind, it is most convenient to report the acidity of the water as equivalent to so much sulphuric acid.
Dissolved Oxygen.—For the gasometric method of analysing for dissolved oxygen, and for the Schützenberger's volumetric method, the student is referred to Sutton's "Volumetric Analysis." The following is an easy method of estimating the free oxygen in a water:—Take 20 c.c. of a stannous chloride solution (about 20 grams of the salt with 10 c.c. of hydrochloric acid to the litre); add 10 c.c. of hydrochloric acid, and titrate in an atmosphere of carbon dioxide with standard permanganate of potassium solution (made by dissolving 1.975 gram of the salt in 1 litre of water: 1 c.c. equals 0.5 milligram of oxygen). A similar titration is made with the addition of 100 c.c. of the water to be tested. Less permanganate will be required in the second titration, according to the amount of oxygen in the water; and the difference, multiplied by 0.5, will give the weight of the oxygen in milligrams. Small quantities of nitrates do not interfere.
In reporting the results of the analysis, it is customary to combine the acids and bases found on some such principle as the following:—The sulphuric oxide is calculated as combined with the potash, and reported as potassic sulphate (K2SO4); the balance of the sulphuric oxide is then apportioned to the soda, and reported as sulphate of soda (Na2SO4); if any is still left, it is reported as calcium sulphate (CaSO4), and after that as magnesic sulphate (MgSO4). When the sulphuric oxide has been satisfied, the chlorine is distributed, taking the bases in the same order, then the nitric pentoxide, and lastly the carbon dioxide. But any method for thus combining the bases and acids must be arbitrary and inaccurate. It is extremely improbable that any[Pg 357] simple statement can represent the manner in which the bases and acids are distributed whilst in solution; and, since different chemists are not agreed as to any one system, it is better to give up the attempt, and simply state the results of the analysis. This has only one inconvenience. The bases are represented as oxides; and, since some of them are present as chlorides, the sum total of the analysis will be in excess of the actual amount present by the weight of the oxygen equivalent to the chlorine present as chloride. The following is an example of such a statement:—
| Parts per 100,000. | |
| Total solids, dried at 100° C. | 28.73 |
| Chlorine | 1.70 |
| Nitrogen as nitrate | 0.03 |
| Ammonia | 0.001 |
| Albuminoid ammonia | 0.004 |
| "Oxygen consumed" in 4 hours | 0.01 |
The solids were made up as under:—
| Per 100,000 of the Water. | |
| Potash | 0.38 |
| Soda | 2.01 |
| Magnesia | 1.44 |
| Lime | 10.55 |
| Ferric oxide | 0.01 |
| Silica | 0.30 |
| Sulphuric oxide | 3.69 |
| Nitrogen pentoxide | 0.11 |
| Carbon dioxide | 8.38 |
| Chlorine | 1.70 |
| Volatile and organic matter | 0.66 |
| ——— | |
| 29.23 | |
| Less oxygen equivalent to chlorine found | 0.39 |
| ——— | |
| 28.84 |
For the preparation of distilled water, the apparatus shown in fig. 64 is convenient for laboratory use. It consists of a copper retort heated by a ring gas-burner, and connected with a worm-condenser.
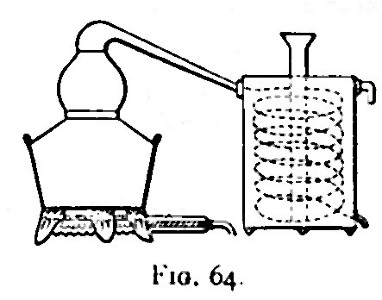
A mineral, on analysis, gave the following results:—Water, 44.94 per cent.; sulphuric oxide, 28.72 per cent.; ferrous iron, 13.92 per cent.; ferric iron, 0.35 per cent.; copper, 6.1 per cent. The mineral was soluble in water, and showed nothing else on testing. How would you report the analysis? Calculate the formula for the salt.[Pg 358]
There is a group of closely allied elements to which the name halogen (salt-producer) has been given. It comprises chlorine, bromine, iodine, and fluorine. These elements combine directly with metals, forming as many series of salts (chlorides, bromides, iodides, and fluorides), corresponding to the respective oxides, but differing in their formulæ by having two atoms of the halogen in the place of one atom of oxygen. For example, ferrous oxide is FeO and ferrous chloride is FeCl2, and, again, ferric oxide is Fe2O3, whilst ferric chloride is Fe2Cl6. These salts differ from the carbonates, nitrates, &c., in containing no oxygen. Consequently, it is incorrect to speak of such compounds as chloride of potash, fluoride of lime, &c., since potash and lime are oxides. It is important to bear this in mind in reporting analyses in which determinations have been made, say, of chlorine, magnesia, and potash, or of fluorine, silica, and alumina. It is necessary in all such cases to deduct from the total an amount of oxygen equivalent to the halogen found, except, of course, where the base has been determined and recorded as metal. Compounds containing oxides and fluorides, &c., do not lend themselves to the method of determining the halogen by difference. For example, topaz, which, according to Dana, has the formula Al2SiO4F2, would yield in the ordinary course of analysis—
| Alumina | 55.4% |
| Silica | 32.6 |
| Fluorine | 20.6 |
| ——— | |
| 108.6 |
The oxygen equivalent to 20.6 per cent. fluorine may be found by multiplying the percentage of fluorine by 0.421; it is 8.7 per cent., and must be deducted. The analysis would then be reported thus:—
| Alumina | 55.4% |
| Silica | 32.6 |
| Fluorine | 20.6 |
| ——— | |
| 108.6 | |
| Less oxygen equivalent to fluorine | 8.7 |
| ——— | |
| 99.9 |
Take as an illustration the following actual analysis by F.W. Clarke and J.S. Diller:[Pg 359]—
| Alumina | 57.38% |
| Silica | 31.92 |
| Fluorine | 16.99 |
| Potash | 0.15 |
| Soda | 1.33 |
| Water | 0.20 |
| ——— | |
| 107.97 | |
| Deduct oxygen equivalent | 7.16 |
| ——— | |
| 100.81 |
In calculating the factor for the "oxygen equivalent," divide the weight of one atom of oxygen (16) by the weight of two atoms of the halogen; for example, with chlorine it would be 16/71 or 0.2253; with bromine, 16/160 or 0.1000; with iodine, 16/254 or 0.063; and with fluorine, 16/38 or 0.421.
Chlorine occurs in nature chiefly combined with sodium, as halite or rock salt (NaCl). With potassium it forms sylvine (KCl), and, together with magnesium, carnallite (KCl.MgCl2.6H2O). Of the metalliferous minerals containing chlorine, kerargyrite, or horn silver (AgCl), and atacamite, an oxychloride of copper (CuCl2.3Cu(HO)2.) are the most important. Apatite (phosphate of lime) and pyromorphite (phosphate of lead) contain a considerable amount of it. Chlorine is a gas of a greenish colour, possessing a characteristic odour, and moderately soluble in water. It does not occur native, and is generally prepared by the action of an oxidising agent on hydrochloric acid. It combines directly with metals at the ordinary temperature (even with platinum and gold), forming chlorides, which (except in the case of silver) are soluble.
It is important in metallurgy, because of the extensive use of it in extracting gold by "chloridising" processes. It is also used in refining gold.
Detection.—Compounds containing the oxides of chlorine are not found in nature, because of the readiness with which they lose oxygen. By reduction they yield a chloride; the form in which chlorine is met with in minerals. In testing, the compound supposed to contain a chloride is boiled with water, or, in some cases, dilute nitric acid. To the clear solution containing nitric acid a few drops of nitrate of silver solution are added. If, on shaking, a white curdy precipitate, soluble in ammonia, separates out, it is sufficiently satisfactory evidence of the presence of chlorides.[Pg 360]
Solution and Separation.—The chlorides are generally soluble in water, and are got into solution by extracting with warm dilute nitric acid. Or, if insoluble, the substance is fused with carbonate of soda, extracted with water, and the filtrate acidified with nitric acid. For the determination, it is not necessary to obtain the solution of the chloride free from other acids or metals. If tin, antimony, mercury, or platinum is present, it is best to separate by means of sulphuretted hydrogen. The chloride is determined in the solution after removal of the excess of the gas. Where traces of chlorides are being looked for, a blank experiment is made to determine the quantity introduced with the reagents. One hundred c.c. of ordinary water contains from 1 to 3 milligrams of chlorine. On the addition of nitrate of silver to the nitric acid solution, chloride of silver separates out. This is free from other substances, except, perhaps, bromide and iodide.
Freely mix the solution containing the chloride with dilute nitric acid, filter (if necessary), and treat with nitrate of silver. Heat nearly to boiling, and, when the precipitate has settled, filter, and wash with hot distilled water. Dry, and transfer to a weighed Berlin crucible. Burn the filter-paper separately, and convert any reduced silver into chloride by alternate treatment with drops of nitric and of hydrochloric acid. Add the main portion to this, and heat cautiously till the edges of the mass show signs of fusing (about 260°). Cool in the desiccator and weigh. The substance is chloride of silver (AgCl), and contains 24.73 per cent. of chlorine.
The precipitated chloride is filtered and washed as soon as possible after settling, since on exposure to light it becomes purple, and loses a small amount of chlorine.
There are several volumetric methods; but that based on the precipitation of silver chloride in neutral solution, by means of a standard solution of silver nitrate (using potassium chromate as indicator), is preferred. Silver chromate is a red-coloured salt; and, when silver nitrate is added to a solution containing both chloride and chromate, the development of the red colour marks off sharply the point at which the chloride is used up. Silver chromate is decomposed and consequently decolorised by solution of any chloride. The solution for this method must be neutral,[Pg 361] since free acid prevents the formation of the red silver chromate. If not already neutral, it is neutralised by titrating cautiously with a solution of soda. In a neutral solution, other substances (such as phosphates and arsenates) also yield a precipitate with a solution of nitrate of silver; and will count as chloride if they are not removed.
The Standard Solution of Nitrate of Silver is made by dissolving 23.94 grams of the salt (AgNO3) in distilled water, and diluting to 1 litre; 100 c.c. are equal to 0.5 gram of chlorine.
The indicator is made by adding silver nitrate to a strong neutral solution of yellow chromate of potash (K2CrO4), till a permanent red precipitate is formed. The solution is allowed to settle, and the clear liquid decanted into a stoppered bottle labelled "chromate indicator for chlorine."
Standardise the silver nitrate by weighing up 0.5 gram of pure sodium chloride (or potassium chloride). Transfer to a flask and dissolve in distilled water; dilute to 100 c.c. Fill an ordinary burette with the standard silver solution, and (after adjusting) run into the flask a quantity sufficient to throw down the greater part of the chlorine. Add a few drops of the chromate indicator and continue the addition of the silver nitrate until the yellow colour of the solution becomes permanently tinted red, after shaking. This shows that the chlorine is all precipitated, and that the chromate is beginning to come down. The further addition of a couple of drops of the silver solution will cause a marked difference in the tint. Read off the quantity run in, and calculate the standard. One gram of sodium chloride contains 0.6062 gram of chlorine; and 1 gram of potassium chloride contains 0.4754 gram.
For the determination of small quantities of chloride (a few milligrams), the same method is used; but the standard solution is diluted so that each c.c. is equal to 1 milligram of chlorine; and the chromate indicator is added before titrating. The standard solution is made by measuring off 200 c.c. of the solution described above, and diluting with distilled water to 1 litre.
Bromine closely resembles chlorine in the nature of its compounds. It does not occur free in nature, but is occasionally found in combination with silver as bromargyrite (AgBr) and, together with chloride, in embolite. It mainly occurs as alkaline bromides in certain natural waters. Nearly all the bromine of commerce is derived from the mother liquors of salt-works—i.e.,[Pg 362] the liquors from which the common salt has been crystallised out. Bromine combines directly with the metals, forming a series of salts—the bromides. In ordinary work they are separated with, and (except when specially tested for) counted as, chlorides. They are detected by adding chlorine water to the suspected solution and shaking up with carbon bisulphide. Bromine colours the latter brown.
Iodine does not occur in nature in the free state; and iodides are rare, iodargyrite or iodide of silver (AgI) being the only one which ranks as a mineral species. Iodates are found associated with Chili saltpetre, which is an important source of the element.
Iodine and Iodides are largely used in the laboratory, and have already been frequently referred to. It is used as an oxidising agent in a similar manner as permanganate and bichromate of potash, especially in the determinations of copper, arsenic, antimony, and manganese.
Iodine is not readily soluble in water; but dissolves easily in a concentrated solution of potassium iodide. Its solutions are strongly coloured; a drop of a dilute solution colours a large volume of water decidedly yellow; on the addition of starch paste, this becomes blue. The delicacy of this reaction is taken advantage of in titrations to determine when free iodine is present. The blue colour may be alternately developed and removed by the addition of iodine (or an oxidising agent) and hyposulphite of soda (or some other reducing agent). In decolorising, the solution changes from blue or black to colourless or pale yellow according to circumstances. Sometimes the solution, instead of remaining colourless, gradually develops a blue which recurs in spite of the further addition of the reducing agent. In these cases the conditions of the assay have been departed from, or (and this is more often the case) there is some substance present capable of liberating iodine.
Iodine forms a series of salts—the iodides—resembling in many respects the chlorides. These can be obtained by direct combination of the metals with iodine.
Detection.—Free iodine is best recognised by the violet vapours evolved from the solution on heating, and by the blue or black colour which it strikes on the addition of starch paste. Iodides are detected by boiling with strong solutions of ferric sulphate or chloride. Iodine is liberated, distilled over, and collected. Chlorine also liberates iodine from iodides; and this reaction is frequently made use of in assaying. A process based on this is[Pg 363] described under Manganese. All substances which liberate chlorine on boiling with hydrochloric acid (dioxides, bichromates, permanganates, &c.) are determined in a similar way.
Solution and Separation.—Most iodides are soluble in water or dilute acids. The separation is effected by distilling the substance with solution of ferric sulphate, and collecting the vapour in a dilute solution of sulphurous acid or arsenite of soda. On the completion of the distillation, the iodine will be in the distillate as iodide; and the gravimetric determination is made on this.
To the solution containing the iodine, as iodide, and which is free from chlorides (and bromides), add a little dilute nitric acid and nitrate of silver till no further precipitate is produced. Filter off, wash with hot water, and dry. Clean the filter-paper as much as possible, and burn it. Collect the ash in a weighed porcelain crucible, add the main portion, and heat to incipient fusion; cool, and weigh. The substance is silver iodide, and contains 54.03 per cent. of iodine.
This is for the titration of free iodine, and is practically that which is described under Manganese. The substance to be determined is distilled with ferric sulphate, and the iodine is collected in a solution of potassium iodide, in which it readily dissolves. If flaky crystals separate out in the receiver, more potassium iodide crystals are added. When the distillation is finished, the receiver is disconnected, and its contents washed out into a beaker and titrated with "hypo." The standard solution of "hypo" is made by dissolving 19.58 grams of hyposulphite of soda (Na2S2O3.5H2O) in water and diluting to 1 litre; 100 c.c. are equal to 1 gram of iodine. To standardise the solution, weigh up 0.25 gram of pure iodine in a small beaker. Add 2 or 3 crystals of potassium iodide; cover with water; and, when dissolved, dilute to 50 or 100 c.c. Titrate, and calculate the standard.
Fluorine is frequently met with as calcium fluoride or fluor-spar (CaF2). It occurs less abundantly as cryolite (Na3AlF6), a fluoride of aluminium and sodium, which is used in glass-making. Certain other rarer fluorides are occasionally met with. Fluorine is also[Pg 364] found in apatite, and in some silicates, such as topaz, tourmaline, micas, &c.
Hydrofluoric acid is used for etching glass and opening up silicates. It attacks silica, forming fluoride of silicon (SiF4), which is volatile. Silica is by this means eliminated from other oxides, which, in the presence of sulphuric acid, are fixed. The commercial acid is seldom pure, and generally weak; and the acid itself is dangerously obnoxious. The use of ammonium fluoride (or sodium fluoride) and a mineral acid is more convenient. Determinations of this kind are made in platinum dishes enclosed in lead or copper vessels in a well-ventilated place. Fluor-spar is useful as a flux in dry assaying; it renders slags, which would otherwise be pasty, quite fluid. Fluorides generally are fusible, and impart fusibility to substances with which they form weak compounds. Their fluxing action does not depend on the removal of silicon as fluoride.
Detection.—Fluorides in small quantity are easily overlooked unless specially sought for. In larger amounts they are recognised by the property hydrofluoric acid has of etching glass. A watch-glass is warmed, and a layer of wax is melted over the convex side. When cold, some lines are engraved on the waxed surface with any sharp-pointed instrument. The substance to be tested is powdered; and moistened, in a platinum dish, with sulphuric acid. The watch-glass is filled with cold water and supported over the dish. The dish is then carefully warmed, but not sufficiently to melt the wax. After a minute or two, the glass is taken off, and the wax removed. If the substance contained fluorine, the characters will be found permanently etched on the glass. An equally good, but more rapid, test is to mix the powdered substance with some silica, and to heat the mixture in a test tube with sulphuric acid. Silicon fluoride is evolved, and, if a moistened glass rod is held in the tube, it becomes coated with a white deposit of silica, formed by the decomposition of the silicon fluoride by the water. This is also used as a test for silica; but in this case the substance is mixed with a fluoride, and the experiment must obviously be carried out in a platinum vessel.
Separation and Determination.—The determination of fluorine is difficult. In the case of fluorides free from silicates (such as fluor-spar), it is determined indirectly by decomposing a weighed portion with sulphuric acid, evaporating, igniting, and weighing the residual sulphate. The increase in weight multiplied by 0.655 gives the weight of fluorine.
In the presence of silica this method does not answer, because of the volatilisation of silicon fluoride. In these cases Wöhler[Pg 365] adopted the following plan, which resembles that for the indirect determination of carbon dioxide. Mix the weighed substance in a small flask with powdered silica and sulphuric acid. The mouth of the flask is closed with a cork carrying a tube which is filled, the first half with calcium chloride and the second half with pumice coated with dried copper sulphate. The apparatus is weighed quickly, and then warmed till decomposition is complete. A current of dry air is aspirated for a minute or two; and the apparatus again weighed. The loss in weight gives that of the silicon fluoride (SiF4), which, multiplied by 0.7307, gives the weight of fluorine.
Fresenius uses the same reaction; but collects and weighs the silicon fluoride. The finely powdered and dried substance is mixed with ten or fifteen times its weight of ignited and powdered silica. The mixture is introduced into a small dry flask connected on one side with a series of drying-tubes, and on the other with an empty tube (to condense any sulphuric acid). To this last is joined a drying-tube containing chloride of calcium and anhydrous copper sulphate. This is directly connected with a series of three weighed tubes in which the fluoride of silicon is collected. The last of these is joined to another drying-tube. The first weighed tube contains pumice and cotton wool, moistened with water; the second tube contains soda-lime as well as (in the upper half of the second limb) fused calcium chloride between plugs of wool; the third tube is filled half with soda-lime and half with fused calcium chloride. The distilling-flask containing the substance mixed with silica is charged with 40 or 50 c.c. of sulphuric acid, and placed on the hot plate. Alongside it is placed a similar dry flask containing a thermometer, and the temperature in this is kept at 150° or 160° C. A current of air is sent through the tubes during the operation, which takes from one to three hours for from 0.1 to 1 gram of the substance. A correction is made by deducting 0.001 gram for every hour the dried air has been passed through. The increase in weight of the three tubes gives the weight of the silicon fluoride.
Penfield uses a similar arrangement, but passes his silicon fluoride into an alcoholic solution of potassium chloride. Silica and potassium silico-fluoride are precipitated, and hydrochloric acid is set free.[100] The acid thus liberated is titrated, with a standard solution of alkali, in the alcoholic solution, and from the amount of free acid found the fluorine is calculated. The weight of hydrochloric acid (HCl) found, multiplied by 1.562, gives the weight of the fluorine. With this method of working, fewer U-tubes[Pg 366] are required. The exit tube from the flask is bent so as to form a small V, which is kept cool in water; this is directly connected with the U-tube containing the alcoholic solution of potassium chloride. The flask with the assay is heated for about two hours, and a current of dry air is aspirated throughout the determination. Fluoride of silicon is a gas not easily condensed to a liquid: but is immediately decomposed by water or moist air.
[95] This will require two or three hours to thoroughly complete. It is best to powder the oxide first produced, and recalcine.
[96] No magnetic oxide was formed.
[97] For example:—
CaO + 2HCl = CaCl2 + H2O.
PbO + H2SO4 = PbSO4 + H2O.
MgO + 2HNO3 = Mg(NO3)2 + H2O.
Al2O3 + 6HCl = Al2Cl6 + 3H2O.
Fe2O3 + 3H2SO4 = Fe2(SO4)3 + 3H2O.
[98] Fe2O3: 2FeO:: 0.2877: 0.2589.
[99] 100 c.c. contain 1 gram of sulphuric acid.
[100] 3SiF4 + 4KCl + 2H2O = 2K2SiF6 + SiO2 + 4HCl.
Sulphur occurs native in volcanic districts, and is mined in Sicily, Italy, and California in considerable quantities. Combined with metals (sulphides), it is common in all mineral districts. Iron pyrites (FeS2) is the most abundant source of this element. Sulphates, such as gypsum, are fairly common, but have no value so far as the sulphur in them is concerned. In coal it exists as an impurity, occurring partly as a constituent of organic compounds.
Sulphur, whether free or combined with metals, forms, on burning, sulphurous oxide (SO2), which by the action of oxidising agents and water is converted into sulphuric acid. It forms two oxides, sulphurous (SO2) and sulphuric (SO3), which combine with bases to form sulphites and sulphates. Sulphites are of little importance to the assayer, and are converted into sulphates by the action of nitric acid and other oxidising agents.
The native sulphides, when acted on with hydrochloric acid, give off sulphuretted hydrogen; with nitric acid or aqua regia, sulphates are formed, and more or less sulphur separated.
Sulphur is detected in sulphides by the irritating odour of sulphurous oxide given off on roasting, by the evolution of sulphuretted hydrogen when treated with hydrochloric acid, or by a white precipitate of barium sulphate formed when barium chloride is added to the aqua regia solution.
Dry Assay.—There is no method of general application. Free or native sulphur may be volatilised, condensed, and weighed, but pyrites only gives up a portion of its sulphur when heated in a closed vessel, while most sulphides, and all sulphates, give up none at all.
In the determination of sulphur in brimstone, 10 grams of the substance are taken, placed in a small porcelain dish, heated over a Bunsen burner in a well-ventilated place, and ignited. When the sulphur has been completely burnt off, the residue (which consists chiefly of sand) is collected and weighed. In a separate portion the moisture and arsenic are determined; the amounts of[Pg 368] these are deducted from the loss in the first experiment. The difference, multiplied by 10, gives the percentage of sulphur.
Solution.—All sulphates, excepting those of lead, barium, strontium, and lime, are soluble in water or dilute acid. All sulphides, except cinnabar, are converted into sulphates by the action of nitric acid at a gentle heat; or, better, by the action of a mixture of three volumes of nitric acid and one volume of hydrochloric acid. This last attacks cinnabar as well. With most substances it is difficult to convert the whole of the sulphur into sulphuric acid. The sulphur separates out at first as a dark spongy mass, which (on continued treatment) changes to light-coloured flakes. When the solution becomes concentrated and the temperature rises sufficiently, the sulphur fuses into one or more honey-coloured globules which, owing to the small surface they oppose to the acid, are very slowly oxidised. It is not desirable to assist the formation of these globules; therefore, the temperature is kept as low as possible, and strong nitric acid is used. When such globules form, it is best to allow the solution to cool, when the globules will solidify. They can then be filtered off and picked out from the insoluble residue, dried, weighed, ignited, and again weighed, the loss being counted as sulphur. With iron pyrites this difficulty seldom occurs.
Metallic sulphides when fused with an excess of nitre are completely oxidised. If the ore is rich in sulphur, some inert body (such as sodium chloride, or, better, sodium carbonate) is added to dilute the action. With pure sulphur, the action is so energetic as to cause an explosion, so that care should be taken. With burnt ores (incompletely calcined pyrites), there is sufficient oxide of iron present to prevent too rapid action.
These fusions with nitre are best conducted in a platinum dish covered with a piece of platinum foil. The ore is ground with the nitre to ensure complete mixing. The heat need not be excessive, so that a single Bunsen burner placed beneath the dish will suffice; if the bottom of the dish is seen to be red-hot, it is sufficient. On cooling and extracting with water, the sulphur will pass into solution as potassium sulphate, which is then filtered off from the insoluble oxides of iron, copper, &c. The filtrate, after having been treated with a large excess of hydrochloric acid, evaporated to dryness, and re-dissolved in water, is ready for the determination.
Lead sulphate may be dissolved by boiling with ammonium acetate. The insoluble sulphates of barium, strontium, and lime,[Pg 369] are decomposed by fusing with 4 or 5 times their weight of "fusion mixture." The alkaline sulphates are then dissolved out with water, and filtered off from the insoluble residue. The filtrate is rendered acid with hydrochloric acid.
Separation.—The determination of the sulphuric acid in these solutions by precipitation with barium chloride also serves as a separation; but in hot acid solutions containing copper, and more especially iron salts, the baric sulphate has a strong tendency to carry down amounts of those bodies, varying, no doubt, with the conditions of the precipitation. Boiling hydrochloric acid fails to completely extract them. Moreover, the use of hot concentrated hydrochloric acid causes a loss by dissolving barium sulphate. Nitric acid and nitrates must be decomposed by prolonged boiling and evaporation with hydrochloric acid. The iron may be removed by adding a slight excess of ammonia to the faintly acid solution, filtering off, and washing the precipitated ferric hydrate with hot water. By slightly acidulating the filtrate with hydrochloric acid, it will be rendered ready for the determination.
This assay is one of those which strikingly shows the necessity of getting the assay solution under proper conditions, in order to obtain satisfactory results. The method has been repeatedly investigated, and the conclusion arrived at, "that it can be correct only by accident." Yet there are many chemists who get good results, and place considerable faith in its accuracy. This can only be due to differences in the manner of working. It is generally understood that nitric acid or nitrates must be absent; and our experience fully confirms this. Precipitations in nitrate solutions are worthless, as the following experiments show. In each experiment the bulk of the solution was 150 c.c. The solutions contained 10 grams of nitre, were freely acid with hydrochloric acid, and were precipitated (while boiling) with slight excess of baric chloride.
| Sulphuric acid | taken | 0.020 | gram | 0.050 | gram | 0.100 | gram |
| " | found | 0.019 | " | 0.047 | " | 0.098 | " |
| " | taken | 0.500 | " | 1.004 | " | 1.000 | " |
| " | found | 0.526 | " | 1.126 | " | 1.126 | " |
All the precipitates were boiled with hydrochloric acid, and thoroughly washed before weighing. The results of some other experiments on this subject are given under "sulphur" in the "examination of commercial copper," page 207.
The solution having been obtained free from nitrates and chlorates[Pg 370] (and containing but little free hydrochloric acid), is largely diluted, heated to boiling, and precipitated with a moderate excess of a solution of chloride of barium (8 parts of the crystallized barium chloride are sufficient for 1 of sulphur). It is allowed to settle for half-an-hour, and then decanted through a filter. The precipitate is shaken up with boiling water, rendered slightly acid, filtered, washed, dried, ignited, and weighed. The ignited precipitate, when pure, is white, and is not decomposed at a red heat; it is barium sulphate (BaSO4), and contains 13.73 per cent. of sulphur, or 34.33 per cent. of sulphuric oxide (SO3).
Determination of Sulphur in Pyrites.—Weigh up half a gram of the dried and powdered sample, and treat with 10 c.c. of a mixture of 3 volumes of nitric acid and 1 volume of hydrochloric acid, occasionally heating. Evaporate to dryness, treat with 5 c.c. of hydrochloric acid, and again evaporate; take up with 1 c.c. of hydrochloric acid and 100 c.c. of hot water, filter through a small filter, and wash. The residue may contain sulphates of lead, barium, or lime; it must be separately examined, if the total sulphur is wanted. The filtrate is heated, and rendered slightly alkaline with ammonia. Filter off the precipitated ferric hydrate through a quick filter, and wash with hot water. If necessary, evaporate the bulk to about 200 c.c., render faintly acid with hydrochloric acid, and add 20 c.c. of solution of barium chloride; allow to stand for half-an-hour, and decant through a filter. Wash with hot water, dry, ignite, and weigh. Pure pyrites contains 53.33 per cent. of sulphur.
This is based upon the easy conversion of all sulphur compounds into sulphates by fusion with nitre or by oxidation with nitric acid; and on the determination of the sulphate formed by titration in an acetic acid solution with baric chloride.[101] The finishing point is determined by filtering off portions of the assay solution, and testing with sulphuric acid. A slight excess of baric chloride will cause a precipitate.
The process may be divided into—(1) the preparation of the solution, and (2) the titration.
Preparation of the Solution.—Weigh up from 1 to 5 grams of the dried and powdered substance, and mix intimately with 4 grams of powdered nitre; clean out the mortar with another gram of nitre, and add this as a cover. Heat in a platinum crucible for fifteen minutes at a low temperature; cool, and extract[Pg 371] with water in an evaporating-dish about 9 inches across, and holding 700 or 800 c.c. Add 10 grams of sodium acetate and 10 c.c. of acetic acid, and dilute to half a litre. Boil. The solution is ready for titrating. Substances which lose sulphur on heating (such as pyrites) are thus treated:—Weigh up 1 gram, and evaporate nearly to dryness with 10 c.c. each of nitric and hydrochloric acids. Take up with 10 c.c. of hydrochloric acid, and again boil down to a small bulk; dilute and transfer to a 9-inch evaporating-dish; add 10 grams of sodium acetate and 5 c.c. of acetic acid, dilute to half a litre, and boil. The solution is ready for titrating. Sulphates may be dissolved up in the dish itself with the help of a c.c. or so of hydrochloric acid; sodium acetate and acetic acid are then added; and, after dilution and boiling, the solutions are at once titrated.
The solution before titration must contain no free mineral acid, but 5 or 10 c.c. of acetic acid should be present. It must contain 10 grams of sodium acetate, or sufficient to convert any free mineral acid into its corresponding sodic salt; or, if chlorides, nitrates or sulphates of the metals are present, sufficient to decompose them. If a precipitation occurs, as is the case with ferric salts, &c., the solution is titrated with the precipitate in it.
The Titration.—The standard solution of barium chloride is made by dissolving 76.25 grams of the crystallized salt (BaCl2.2H2O) in distilled water, and diluting to 1 litre. 100 c.c. will equal 1 gram of sulphur. As indicator, use dilute sulphuric acid. The strength of the solution may be checked by the titration of 5 grams of ferrous sulphate (oxidized with permanganate of potassium or a few drops of nitric acid), which should require 57.5 c.c. of the barium chloride solution; or any pure sulphate of known composition can be used; anhydrous salts should be preferred.
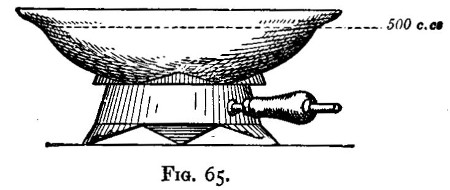
Fill an ordinary 100 c.c. burette with the solution of barium chloride. The evaporating dish containing the assay solution is placed on a round burner (as shown in fig. 65), and the solution is kept steadily boiling. An ordinary Bunsen-burner flame will cause bumping, and should not be used. Run in the standard[Pg 372] solution in quantity known to be insufficient; then withdraw a portion of about 2 c.c., with a pipette, and filter through a fine filter-paper into a test tube. Run in another 0.5 c.c. of the standard solution, and withdraw and filter into a test tube another portion of 2 c.c.; and continue this operation until half-a-dozen or more portions have been drawn off. The test tubes should be arranged in order in a stand resting on a piece of paper, so that each test tube representing 0.5 c.c. of the standard baric chloride may have its value recorded beneath it (fig. 66). Add to each test tube 3 drops of dilute sulphuric acid; that which shows the first appearance of a precipitate marks the point at which the titration is complete. Suppose, for example, that the test tube marked 48.5 c.c. shows no precipitate, while that at 49.0 c.c. shows one, it is evident that the finishing point lies between these readings. With a little practice, one can judge from the appearance of the precipitate in the 49 c.c. tube, whether 1/4 c.c. should be deducted or not.
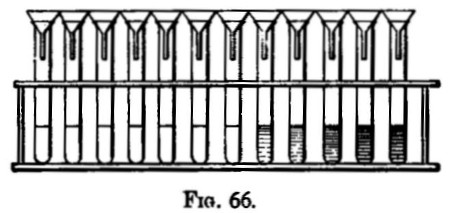
It is better to add dilute sulphuric acid, and to watch for the appearance of a precipitate in the test tube, than to add baric chloride and to look for its non-appearance; besides, baric chloride is much less likely to be present in a test tube as impurity than sulphates are. In this way the chance of error from what are termed "accidental causes" is diminished.
The following experiments show the effect of variation in the conditions of titration:—
Make a standard solution of sulphuric acid by diluting 43.65 grams of sulphuric acid (sp. g. 1.6165) to 1 litre: 100 c.c. will contain 1 gram of sulphur. An equivalent solution may be made by dissolving 100.62 grams of sodium sulphate crystals (Na2SO4.10H2O), or 86.88 grams of ferrous sulphate (FeSO4.7H2O), in water (oxidising the latter), and diluting to 1 litre.
The order in which these experiments are given is that in which they were made in an investigation into the conditions under which the titration could most accurately be effected.
Effect of Hydrochloric and Nitric Acids.—The titrations were performed in the manner already described, but sodic acetate[Pg 373] and acetic acid were absent. Twenty c.c. of the standard solution of sulphuric acid were used.
| Hydrochloric acid present | 0.0 | c.c. | 1.0 | c.c. | 2.0 | c.c. | 5.0 | c.c. |
| "Baric chloride" required | 20.0 | " | 20.0 | " | 19.7 | " | 12.5 | " |
| Nitric acid present | 0.0 | c.c. | 1.0 | c.c. | 2.0 | c.c. | 5.0 | c.c. |
| "Baric chloride" required | 20.0 | " | 19.5 | " | 18.0 | " | 10.0 | " |
These show clearly the interference of free mineral acids, although very dilute hydrochloric acid (1 c.c. in 500 of water) has no effect.
Effect of Acetic and Citric Acids.—A similar series of experiments with these acids gave the following results:—
| Acetic acid present | 0.0 | c.c. | 5.0 | c.c. 50.0 | c.c. 100.0 | c.c. | ||
| "Baric chloride" required | 20.0 | " | 20.0 | " | 20.0 | " | 20.0 | " |
| Citric acid present | 0 gram | 1 gram | 5 grams |
| "Baric chloride" required | 20.0 c.c. | 20.0 c.c. | 20.0 c.c. |
These acids do not interfere.
Effect of Sodic Acetate and Acetic Acid.—In each of these experiments 5 c.c. of acetic acid was present.
| Sodium acetate added | 0 gram | 1 gram | 10 grams | 50 grams |
| "Baric chloride" required | 20.0 c.c. | 20.0 c.c. | 20.0 c.c. | 20.0 c.c. |
As sodic acetate and acetic acid did not interfere, it became desirable to make some experiments on the finishing point. The first object sought for was the smallest amount of the standard baric chloride in 500 c.c. of water, required to give an indication when tested in the manner already described.
| Conditions of Assay Solution. | Baric Chloride required. |
| Water only | 0.05 c.c. |
| With 10 grams of sodium acetate and 5 c.c. of acetic acid | 0.05 " |
| The same with 5 grams of nitre | 0.10 " |
| Like the last, but with 5 grams of salt instead of nitre | 0.10 " |
These show that as small an amount of baric chloride solution as is equal to only 0.000002 gram of sulphur in the 2 c.c. of solution tested yields a decided precipitate on the addition of 3 drops of sulphuric acid.
To determine whether the same finishing point is obtained on testing the filtered portions in the test tubes with baric chloride as is obtained on testing with sulphuric acid, a titration was made with 20 c.c. of standard solution of sulphuric acid, together with the usual quantities of sodic acetate and acetic[Pg 374] acid; and two lots of 2 c.c. each were filtered into two sets of test tubes after each addition of the standard baric chloride. To one series 3 drops of baric chloride solution were added, and to the other 3 drops of sulphuric acid. The results were—
| "Baric Chloride" added. | With Dilute Sulphuric Acid. | With Baric Chloride Solution. |
| 19.5 c.c. | Clear | Cloudy |
| 19.75 " | Clear | Cloudy |
| 20.0 " | Finished | Finished |
| 20.25 " | Cloudy | Clear |
| 20.5 " | Cloudy | Clear |
The two methods of testing give the same result. But this balance is disturbed in the presence of much nitre, the indications with baric chloride being disturbed by an opalescence for some c.c. beyond the finishing point. In solutions containing free hydrochloric or nitric acid, a precipitate is obtained with either baric chloride or sulphuric acid.
Effect of Varying Sulphur.—In these and the subsequent experiments the titrations were performed in the presence of 10 grams of sodic acetate and 10 c.c. of acetic acid in the manner already described.
| Standard sulphuric acid used | 5.0 c.c. | 10.0 c.c. | 20.0 c.c. | 50.0 c.c. | 100.0 c.c. |
| "Baric chloride" required | 5.0 " | 10.0 " | 20.0 " | 50.0 " | 100.0 " |
Effect of Varying Temperature.—With 5 c.c. of standard sulphuric acid titrated at 15° C., 5 c.c. of baric chloride were required; but with larger quantities the results were altogether unsatisfactory when titrated cold.
Effect of Varying Bulk.—
| Bulk | 100.0 c.c. | 200.0 c.c. | 500.0 c.c. | 1000.0 c.c. |
| "Baric chloride" required | 20.0 " | 20.0 " | 20.0 " | 20.5 " |
Considerable variation in bulk has no effect, but 500 c.c. is the most convenient volume to work with. It is well to occasionally replace the water boiled off during titration.
Effect of Foreign Salts.—In all these experiments 20 c.c. of "sulphuric acid" were used, and the titration was performed in the ordinary way.
| Sodic chloride added | 0 gram | 5 grams | 10 grams | |
| "Baric chloride" required | 20.0 c.c. | 20.0 c.c. | 19.7 c.c. | |
| Ammonic chloride added | 0 gram | 5 grams | 10 grams | |
| "Baric chloride" required | 20.0 c.c. | 20.0 c.c. | 19.5 c.c. | |
| Calcic chloride added | 0 gram | 1 gram | 2 grams | 5 grams |
| "Baric chloride" required | 20.0 c.c. | 20.0 c.c. | 19.2 c.c. | 19.0 c.c. |
| Zinc chloride added | 0 gram | 1 gram | 3 grams | 5 grams |
| "Baric chloride" required | 20.0 c.c. | 20.0 c.c. | 20.0 c.c. | 20.0 c.c. |
| Ferrous chloride added | 0 gram | 1 gram | 3 grams | 5 grams |
| "Baric chloride" required | 20.0 c.c. | 19.7 c.c. | 19.5 c.c. | 19.0 c.c. |
| Ferric chloride added | 0 gram | 1 gram | 3 grams | 5 grams |
| "Baric chloride" required | 20.0 c.c. | 20.0 c.c. | 20.0 c.c. | 20.0 c.c. |
| Copper chloride added | 0 gram | 1 gram | 3 grams | 5 grams |
| "Baric chloride" required | 20.0 c.c. | 20.0 c.c. | 20.0 c.c. | 20.0 c.c. |
| Potassic Nitrate added | 0 gram | 1 gram | 5 grams | 10 grams |
| "Baric chloride" required | 20.0 c.c. | 20.0 c.c. | 20.0 c.c. | 19.0 c.c. |
| Potassic Nitrite added | 0 gram | 1 gram | 5 grams | |
| "Baric chloride" required | 20.0 c.c. | 20.0 c.c. | 20.0 c.c. | |
| Sodic phosphate added | 0 gram | 1 gram | ||
| "Baric chloride" required | 20.0 c.c. | 22.5 c.c. | ||
| Sodic arsenate added | 0 gram | 1 gram | ||
| "Baric chloride" required | 20.0 c.c. | 20.5 c.c. |
In the absence of ferric salts, phosphates and arsenates count as sulphur.
In two series of experiments for determining the effect of varying amounts of sulphur in the form of ferrous sulphate, we obtained the following results:—In the first series the assay solution was prepared in the manner we have described for Pyrites; and in the second series, by fusion with nitre.
| Sulphur added | 0.050 gram | 0.100 gram | 0.200 gram | ||
| "Baric chloride" | required | (1) | 5.0 c.c. | 10.0 c.c. | 20.0 c.c. |
| " | " | (2) | 4.7 " | 10.0 " | 20.0 " |
| Sulphur added | 0.500 gram | 1.000 gram | |||
| "Baric chloride" | required | (1) | 50.0 c.c. | 100.0 c.c. | |
| " | " | (2) | 50.0 " | 100.0 " |
More than 5 grams of nitre must not be used in an assay; and, since the requisite amount of nitre considerably exceeds that sufficient to oxidise the sulphur, not more than 0.5 gram of unoxidised sulphur should be present in the portion of the sample weighed up for determination. When the amount of sulphur present is not known within reasonable limits, the test portions may be tried with a drop of baric chloride solution instead of sulphuric acid, so that the diminishing quantity of precipitate may give warning of an approach to the finishing point.
Determination of Sulphur in Blende.—Weigh up 1 gram[Pg 376] of dried and powdered blende, and mix and fuse with 5 grams of nitre in the manner described. Place the dish and its contents in the titrating-dish, extract with water, add 10 grams of sodium acetate and 10 c.c. of acetic acid, remove and wash the platinum-dish, and dilute to 500 c.c.; boil and titrate. In the example, duplicate determinations required (a) 32.0 c.c., (b) 32.25 c.c., giving an average of 32.1 per cent. of sulphur.
Determination of Sulphur in Chalcopyrite (Yellow Copper Ore).—Take 1 gram of the finely-powdered sample, and 5 grams of nitre. Sprinkle a little of the nitre in a small Wedgwood mortar, place the ore on it, and cover with 2 or 3 grams more of the nitre. Rub up together, and transfer to a small porcelain dish; clean out the mortar with the rest of the nitre, and add to the contents of the dish. Cover with a piece of platinum foil, and heat gently with a Bunsen burner till the nitre melts and the stuff shows signs of deflagrating; remove the heat, and allow the action to go on by itself for a minute or so, then heat over the Bunsen burner for 10 minutes. Cool; transfer the whole to the titrating-dish; boil with 500 c.c. of water; remove the small dish and foil; add sodic acetate and acetic acid, and titrate.
For example, 1 gram required 34.5 c.c. of "barium chloride" (standard = 1.005 gram S), which is equivalent to 34.7 per cent. sulphur. The theoretical percentage is 34.8.
Determination of Sulphur in Chalcocite (Grey Copper Ore).—Proceed as in the last experiment but, since the action with nitre is more moderate, no special precautions need be taken on heating. A platinum dish may be used.
An example which was heated for 30 minutes required 20.5 c.c. of the barium chloride solution. This is equivalent to 20.6 per cent. of sulphur. The theoretical yield is 20.2 per cent.
Determination of Sulphur in Pyrites.—Take 1 gram of the finely-powdered sample, cover with 10 c.c. of nitric acid, and, when action has ceased, evaporate to a small bulk. Add 3 or 4 c.c. of hydrochloric acid, and again evaporate to a paste. Take up with 1 or 2 c.c. of dilute hydrochloric acid, dilute with water, transfer to a titrating-dish, add 10 grams of sodic acetate and 5 c.c. of acetic acid, and dilute with water to 500 c.c. Boil and titrate.
An example with 1 gram of a pure crystallized pyrites required 52.7 c.c. of the barium chloride solution, which is equivalent to 53.0 per cent. of sulphur. Theory requires 53.3 per cent. of sulphur.
Determination of Sulphur in Mispickel.—Take 1 gram of the powdered ore and evaporate with 10 c.c. of nitric acid, and take up with 3 or 4 c.c. of hydrochloric acid. If any globules of[Pg 377] sulphur remain, again evaporate with nitric acid. Dilute, and transfer to the titrating-dish. Add 10 grams of sodic acetate, dilute with water, boil, and titrate. The mispickel carries (according to theory) exactly sufficient iron to precipitate the arsenic as ferric arsenate in an acetic acid solution, so no more iron need be added. The ferric arsenate will separate out as a yellowish-white flocculent precipitate.
An example required, in duplicate experiment, 18.5 c.c. and 18.7 c.c. of barium chloride, equivalent to 18.7 per cent. of sulphur. The formula, FeS2.FeAs2, requires 19.6 percent., but the sulphur generally varies considerably from this amount.
Determination of Sulphur in Burnt Ores.—Take 5 grams of the dried and powdered ore, and rub up with 4 grams of nitre; transfer to the platinum-dish; clean out the mortar with another gram of nitre, and add this as a cover. Heat, and extract with water as before; add the sodium acetate and acetic acid; and titrate. Burnt ores carry from 2.5 to 5 per cent. of sulphur. A series of four determinations gave:—
| "Baric Chloride" Required. | Percentage of Sulphur. | Gravimetric Results. |
| 12.6 c.c. | 2.52 % | 2.45 % |
| 29.9 " | 5.98 " | 5.84 " |
| 18.1 " | 3.62 " | 3.53 " |
| 22.0 " | 4.40 " | 4.43 " |
For ores carrying less than 1 per cent. of sulphur, take 10 grams for the assay.
Determination of Sulphuric Oxide (SO3) in Sulphates.—When the sulphur exists in the sample received by the assayer in an oxidised state as sulphate, it is usual to report it in terms of sulphuric oxide (SO3). In this case, the metal must also be reported as oxide. For example, an analysis of copper sulphate would be thus reported:—
| Oxide of copper (CuO) | 31.8 % |
| Sulphuric oxide (SO3) | 32.1 " |
| Water | 36.1 " |
| ——— | |
| 100.0 |
The percentage of sulphur multiplied by 2.5 gives the percentage of sulphuric oxide. Thus a sample of copper sulphate containing 12.85 per cent. of sulphur will contain 12.85 × 2.5 or 32.12 per cent. of sulphuric oxide.
In minerals and metallurgical products, it is common to find the sulphur in both conditions—i.e., as sulphate and sulphide. Generally in these the percentage of sulphur only is wanted; but this will depend entirely on commercial requirements, and not on[Pg 378] the fancy of the assayer. Soluble sulphates are determined separately by extracting with small quantities of cold water, so as to avoid the separation of basic sulphates, or, if the sulphides present are not at the same time attacked, by dilute hydrochloric acid. Lead sulphate may be extracted by boiling with ammonic acetate; whilst barium, strontium, and, perhaps, calcium sulphate, will be mainly found in the residue insoluble in acids.
Weigh up from 2 to 5 grams of the material according to the amount of sulphur judged to be present, and dissolve them in the titrating-dish with 1 c.c. of hydrochloric acid and 50 c.c. of water. Add 10 grams of sodic acetate, and 10 c.c. of acetic acid; dilute, boil, and titrate. In the case of ferric salts, half the quantity of acetic acid will be better, as then the ferric iron will be precipitated, and a colourless solution will be left, in which the end reaction is more readily distinguished.
Determined in this way, 5 gram samples of the following salts gave the results indicated below:—
| Salt. | "Barium Chloride" Required. | Sulphuric Oxide. |
| Copper sulphate | 64.25 c.c. | 32.12 % |
| Magnesium sulphate | 65.25 " | 32.62 " |
| Zinc sulphate | 56.25 " | 28.12 " |
| Ferrous sulphate | 58.25 " | 29.12 " |
| Sodium sulphate | 51.25 " | 25.60 " |
Determination of Sulphuric Oxide in Barytes (Heavy spar).—Fuse 2 grams of the powdered mineral with 5 grams of "fusion mixture" for five minutes; and, when cold, extract with water. Filter, acidulate the filtrate with an excess of 10 c.c. of acetic acid, dilute, boil, and titrate. For example, a transparent crystallised sample required 27.0 c.c. of barium chloride, which is equivalent to 13.6 per cent. of sulphur, or 34.0 per cent. of sulphuric oxide. Theory requires 34.3 per cent. of the latter. Since both carbonate of soda and potash are liable to contain sulphates, a blank determination should be made on 5 grams of the "fusion mixture," and the amount found be deducted from that got in the assay.
1. The price of sulphur in an ore being 4-1/2d. per unit in the northern markets, what would be the price of a ton of ore containing 49 per cent. of sulphur? What would be the effect on the price of an error of 0.25 per cent. in the assay?
2. Pyrites carries 50 per cent. of sulphur, and on calcining yields 70 per cent. of its weight of burnt ore. Supposing the burnt ore carries 3.5 per cent. of sulphur, what proportion of the sulphur will have been removed in the calcining?[Pg 379]
3. How would blende compare with pyrites as a source of sulphur for sulphuric acid making?
4. How would you determine the percentage of sulphuric oxide in a sample of gypsum? What is sulphuric oxide, and what relation does it bear to sulphur?
5. A mineral contains 20.7 per cent. of water, 32.4 per cent. of lime, and 18.6 per cent. of sulphur. What is its probable composition? What experiment would you try to determine the accuracy of your conclusion?
occurs in nature combined with copper, mercury, and lead, in certain rare minerals. In small quantities it is found in many ores. It is detected in solution by the red precipitate produced on boiling the acid solution with sodium sulphite. This reaction is used for its determination.
Solution.—The solution is effected by boiling with nitric acid or aqua regia, or by fusing with nitre. To separate the selenium, the solution is evaporated with an excess of hydrochloric acid and a little sodium or potassium chloride. This destroys any nitric acid that may be present, and reduces selenic acid (H2SeO4) to selenious (H2SeO3). The solution is diluted with water, and treated with a solution of sulphite of soda. It is warmed, and at last boiled. The selenium separates as a red precipitate, which (on boiling) becomes denser and black. It is collected on a weighed filter, washed with hot water, dried at 100° C., and weighed as pure selenium.
Selenium can be precipitated with sulphuretted hydrogen as a sulphide, which is readily soluble in ammonium sulphide. This sulphide may be oxidised with hydrochloric acid and chlorate of potash; and the selenium separated in the manner described.
Tellurium occurs in nature, native, and in combination with gold, silver, bismuth and lead. It is sometimes met with in assaying gold ores. It may be detected by the purple colour it imparts to strong sulphuric acid when dissolved in the cold, and by the black precipitate of metallic tellurium which its solutions yield on treatment with a reducing agent. Telluric acid is reduced to tellurous (with evolution of chlorine) on boiling with hydrochloric acid.
Solution is effected by boiling with aqua regia, or by fusing with nitre and sodium carbonate.
Separation.—Tellurium closely resembles selenium in its reactions. It is separated and determined in the same way. Like[Pg 380] it, it forms a sulphide soluble in ammonium sulphide. It is distinguished from selenium by the insolubility, in a solution of cyanide of potassium, of the metal precipitated by sodium sulphite; whereas selenium dissolves, forming a soluble potassic seleno-cyanide.[102]
For the determination, solution is effected by fusing with nitre and sodium carbonate, dissolving out the tellurate of potash with water, and boiling with hydrochloric acid. Tellurous compounds are formed, with evolution of chlorine; and the solution, on treating with a reducing agent (such as sulphurous acid or stannous chloride), yields metallic tellurium; which is washed, dried at 100° C., and weighed.
The chief source of the arsenic of commerce is arsenical pyrites, or mispickel, which contains about 45 per cent. of arsenic (As). Arsenic also occurs as a constituent of several comparatively rare minerals; and, as an impurity, it is very widely distributed. White arsenic is an oxide of arsenic, and is obtained by roasting arsenical ores, and refining the material (crude arsenic), which condenses in the flues. Arsenic itself is volatile, and many of its compounds have the same property. It forms two well-defined series of salts, corresponding to the oxides: arsenious oxide (As2O3), and arsenic oxide (As2O5). These combine with bases to form arsenites and arsenates respectively. Boiling with nitric acid converts the lower into the higher oxide; and powerful reducing-agents, such as cuprous chloride, have the opposite effect.
Arsenic may be detected by dissolving the substance in hydrochloric acid, or in aqua regia (avoiding an excess of nitric acid), and adding a little of this solution to the contents of a small flask in which hydrogen is being made by the action of zinc and hydrochloric acid. The ignited jet of hydrogen assumes a blue colour if arsenic is present, and a cold porcelain dish held in the flame (fig. 67) becomes coated with a dark deposit of metallic arsenic. Antimony produces a similar effect, but is distinguished by the insolubility of its deposit in a cold solution of bleaching-powder.
Arsenites are distinguished by the volatility of the chloride; by decolorising a solution of permanganate of potassium, and by immediately giving a yellow precipitate with sulphuretted hydrogen. Arsenates are distinguished (after converting into soda salts by[Pg 382] boiling with carbonate of soda and neutralising) by giving with nitrate of silver a red precipitate, and with "magnesia mixture" a white crystalline one.
Dry Assay.—There is no dry assay which is trustworthy. The following method is sometimes used to find the proportion of arsenious oxide in "crude arsenic":—Weigh up 5 grams of the dried sample, and place them in a clean dry test-tube about 6 inches long. Tie a small filter-paper over the mouth of the tube, so as to prevent air-currents. Heat the tube cautiously so as to sublime off the white arsenic into the upper part of the tube. Cut off the bottom of the test-tube by wetting whilst hot. Scrape out the arsenic and weigh it. The weight gives an approximate idea of the quantity, and the colour of the quality, of the white arsenic obtainable from the sample. Some workers (sellers) weigh the residue, and determine the white arsenic by difference. In determining the percentage of moisture in these samples, the substance is dried on a water-bath or in a water-oven.
Solution.—Where, as in crude arsenic, the substance is arsenious oxide (As2O3) mixed with impurities, the arsenic is best got into solution by warming with caustic soda, and neutralising the excess with hydrochloric acid; it will be present as sodium arsenite. Metals and alloys are acted on by means of nitric acid; or the arsenic may be at the same time dissolved and separated by distilling with a strongly-acid solution of ferric chloride, in the way described under Volumetric Methods.
With minerals, mattes, &c., solution is thus effected:—The finely-powdered substance is mixed (in a large platinum or porcelain crucible) with from six to ten times its weight of a mixture of equal parts of carbonate of soda and nitre. The mass is then heated gradually to fusion, and kept for a few minutes in that state. When cold, it is extracted with warm water, and filtered from the insoluble residue. The solution, acidified with nitric acid and boiled, contains the arsenic as sodium arsenate. With mispickel, and those substances which easily give off arsenic on heating, the substance is first treated with nitric acid, evaporated to dryness, and then the residue is treated in the way just described.
When the arsenic is present as arsenite or arsenide, distillation with an acid solution of ferric chloride will give the whole of the arsenic in the distillate free from any metal except, perhaps, tin as stannic chloride. With arsenates, dissolve the substance in acid and then add an excess of soda. Pass sulphuretted hydrogen into the solution; warm, and filter. Acidulate the[Pg 383] filtrate, and pass sulphuretted hydrogen. Decant off the liquid through a filter, and digest the precipitate with ammonic carbonate; filter, and re-precipitate with hydrochloric acid and sulphuretted hydrogen. Allow to stand in a warm place, and filter off the yellow sulphide of arsenic. Wash it into a beaker, clean the filter-paper (if necessary) with a drop or two of dilute ammonia; evaporate with 10 c.c. of dilute nitric acid to a small bulk; dilute; and filter off the globules of sulphur. The filtrate contains the arsenic as arsenic acid.
Having got the arsenic into solution as arsenic acid, and in a volume not much exceeding 50 c.c., add about 20 c.c. of dilute ammonia and 20 c.c. of "magnesia mixture." Stir with a glass rod, and allow to settle overnight. Filter, and wash with dilute ammonia, avoiding the use of large quantities of wash water. Dry, transfer the precipitate to a Berlin crucible, and clean the filter-paper thoroughly. Burn this paper carefully and completely; and add the ash to the contents of the crucible, together with 4 or 5 drops of nitric acid. Evaporate with a Bunsen burner, and slowly ignite, finishing off with the blow-pipe or muffle. Cool, and weigh. The ignited precipitate is pyrarsenate of magnesia (Mg2As2O7), and contains 48.4 per cent. of arsenic (As).
Instead of igniting the precipitate with nitric acid, it may be collected on a weighed filter-paper, dried at 100° C., and weighed as ammonic-magnesic arsenate (2AmMgAsO4.H2O), which contains 39.5 per cent. of arsenic. The results in this case are likely to be a little higher. The drying is very tedious, and is likely to leave behind more water than is allowed for in the formula. In a series of determinations in which the arsenic was weighed in both forms, the results were:—
| Ammonic-magnesic Arsenate in grams. | Arsenic in grams. | Magnesium Pyrarsenate in grams. | Arsenic in grams. |
| 0.0080 | 0.0032 | 0.0065 | 0.0031 |
| 0.0400 | 0.0158 | 0.0330 | 0.0160 |
| 0.0799 | 0.0316 | 0.0633 | 0.0306 |
| 0.1600 | 0.0632 | 0.1287 | 0.0623 |
| 0.4000 | 0.1580 | 0.3205 | 0.1551 |
| 0.7990 | 0.3156 | 0.6435 | 0.3114 |
There are two methods: one for determining the arsenic in the lower, and the other in the higher state of oxidation. In the first-mentioned method this is done by titrating with a standard solution of iodine; and in the latter with a solution of uranium acetate. Where the arsenic already exists as arsenious oxide, or where it is most conveniently separated by distillation as arsenious chloride, the iodine method should be used; but when the arsenic is separated as ammonic-magnesic arsenate or as sulphide, the uranium acetate titration should be adopted.
This is based on the fact that sodium arsenite in a solution containing an excess of bicarbonate of soda is indirectly oxidised by iodine to sodium arsenate,[103] and that an excess of iodine may be recognised by the blue colour it strikes with starch. The process is divided into two parts—(1) the preparation of the solution, and (2) the titration.
Preparation of the Solution.—For substances like crude arsenic, in which the arsenic is present as arsenious oxide, the method is as follows:—Take a portion which shall contain from 0.25 to 0.5 gram of the oxide, place in a beaker, and cover with 10 c.c. of sodic hydrate solution; warm till dissolved, put a small piece of litmus paper in the solution, and render acid with dilute hydrochloric acid. Add 2 grams of bicarbonate of soda in solution, filter (if necessary), and dilute to 100 c.c. The solution is now ready for titrating.
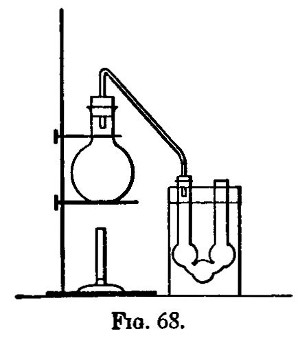
Where the arsenic has to be separated as arsenious chloride, the process is as follows:[104]—Weigh up 1 gram of the finely-powdered ore (metals should be hammered out into a thin foil or be used as filings), and place in a 16-ounce flask provided with a well-fitting cork, and connected with a U-tube, as shown in the drawing (fig. 68). The U-tube should contain 2 or 3 c.c. of water, and is cooled by being[Pg 385] placed in a jar or large beaker of cold water. The water used for cooling should be renewed for each assay.
Pour on the assay in the flask 50 c.c. of a "ferric chloride mixture," made by dissolving 600 grams of calcium chloride and 300 grams of ferric chloride in 600 c.c. of hydrochloric acid, and making up to 1 litre with water.
Firmly cork up the apparatus, and boil over a small Bunsen-burner flame for fifteen or twenty minutes, but avoid evaporating to dryness. Disconnect the flask, and pour away its contents at once to prevent breakage of the flask by their solidification. The arsenic will be condensed in the U-tube, together with the greater part of the hydrochloric acid; transfer the distillate to a beaker washing out the tube two or three times with water; add a small piece of litmus paper; neutralise with ammonia; render faintly acid with dilute hydrochloric acid; add 2 grams of bicarbonate of soda in solution; and dilute to 250 c.c. The solution is now ready for titrating.
The arsenic comes over in the early part of the distillation, as will be seen from the following experiment, made on 1 gram of copper precipitate; in which experiment the distillate was collected in separate portions at equal intervals, and the arsenic in each portion determined:—
| Time Distilling. | Iodine Required. | Equivalent to Arsenic in the Distillate. |
| 5 minutes | 12.0 c.c. | 0.0450 gram |
| 5 " | 0.17 " | 0.0005 " |
| 5 " | 0.0 " | |
| 5 " | 0.0 " | |
| To dryness | 0.0 " |
The volume of each distillate was about 5 c.c.
In this operation the metals are converted into chlorides by the action of ferric chloride, which gives up a part of its chlorine, and becomes reduced to the ferrous salt. The calcium chloride does not enter into the chemical reaction, but raises the temperature at which the solution boils, and is essential for the completion of the distillation.[105] Two experiments with material containing 3.48 per cent. of arsenic gave—(1) with ferric chloride alone, 2.74 per cent.; and (2) with the addition of calcium chloride, 3.48 per cent.
It is always necessary to make a blank determination with 1 gram of electrotype copper, to find out the amount of arsenic in the ferric chloride mixture.[106] Unfortunately, a correction is[Pg 386] always required. This amounts to about 0.15 per cent. of arsenic on each assay, even when the mixture has been purified; and this constitutes the weakness of the method, since, in some cases, the correction is as much as, or even greater than, the percentage to be determined.
The acid distillate containing the arsenious chloride may be left for an hour or so without much fear of oxidation; but it is safer to neutralise and then to add the bicarbonate of soda, as the following experiments show. Several portions of a solution, each having a bulk of 100 c.c., were exposed for varying lengths of time, and the arsenic in each determined.
| Time Exposed. | Acid Solutions. "Iodine" Required. Arsenic Found. | Neutralised Solutions. "Iodine"Required. Arsenic Found. |
| — | 18.2 c.c. = 0.0136 gram | 18.1 c.c. = 0.0136 gram |
| 1 hour | 18.2 " = 0.0136 " | 18.2 " = 0.0136 " |
| 2 hours | 17.7 " = 0.0133 " | 18.0 " = 0.0135 " |
| 4 " | 17.5 " = 0.0131 " | 18.4 " = 0.0138 " |
| 5 " | 17.0 " = 0.0127 " | 18.3 " = 0.0137 " |
The Titration.—Make a standard solution of iodine by weighing up in a beaker 16.933 grams of iodine and 30 grams of potassium iodide in crystals; add a few c.c. of water, and, when dissolved, dilute to 1 litre: 100 c.c. will equal 0.500 gram of arsenic.
A solution of starch similar to that used in the iodide-copper assay will be required. Use 2 c.c. for each assay. Variations in the quantity of starch used do not interfere; but the solution must be freshly prepared, as after seven or eight days it becomes useless.
To standardise the iodine solution, weigh up 0.3 gram of white arsenic; dissolve in caustic soda; neutralise; after acidulating, add 2 grams of bicarbonate of soda and 2 c.c. of the starch solution, and dilute to 200 c.c. with cold water. Fill a burette having a glass stop-cock with the iodine solution, and run it into the solution of arsenic, rapidly at first, and then more cautiously, till a final drop produces a blue colour throughout the solution. Calculate the standard in the usual way. White arsenic contains 75.76 per cent. of arsenic.
The following experiments show the effect of variation in the conditions of the titration:[Pg 387]—
Make a solution of arsenic by dissolving 6.60 grams of white arsenic in 100 c.c. of sodic hydrate solution; render slightly acid with hydrochloric acid; add 10 grains of bicarbonate of soda, and dilute to 1 litre: 100 c.c. will contain 0.50 gram of arsenic.
Effect of Varying Temperature.—The reaction goes on very quickly in the cold, and, since there is no occasion for heating, all titrations should therefore be carried out cold.
Effect of Varying Bulk.—In these experiments, 20 c.c. of arsenic solution were taken, 2 grams of bicarbonate of soda and 2 c.c. of starch solution added, and water supplied to the required bulk. The results were:—
| Bulk | 50.0 c.c. | 100.0 c.c. | 250.0 c.c. | 500.0 c.c. |
| "Iodine" required | 20.0 " | 20.0 " | 20.0 " | 20.0 " |
Considerable variation in bulk does not interfere.
Effect of Varying Bicarbonate of Soda.—This salt must be present in each titration in considerable excess, to prevent the interference of free acid. The bicarbonate must be dissolved without heating, as neutral carbonates should be avoided.
| Bicarbonate added | 1 gram | 2 grams | 5 grams | 10 grams |
| "Iodine" required | 20.1 c.c. | 20.0 c.c. | 20.1 c.c. | 20.0 c.c. |
These results show that large variation in the quantity of bicarbonate has no effect.
Effect of Free Acid.—In these experiments, the arsenic taken, the starch, and the bulk were as before, but no bicarbonate was added. In one case the solution was rendered acid with 5 c.c. of acetic acid, and in the other with 5 c.c. of hydrochloric acid; in both cases the interference was strongly marked, and no satisfactory finishing point could be obtained. This was much more marked with the hydrochloric acid.
Effect of Foreign Salts.—The process for getting the arsenic into solution will exclude all metals except tin, but the solution will be charged with sodium or ammonium salts in the process of neutralising, so that it is only necessary to see if these cause any interference. The alkaline hydrates, including ammonia, are plainly inadmissible, since no free iodine can exist in their presence. Monocarbonates similarly interfere, but to a much less extent; hence the necessity for rendering the assay distinctly acid before adding the bicarbonate of soda.
With 20 c.c. of arsenic solution; and with bulk, soda, and starch as before, the results obtained were:[Pg 388]—
| "Iodine" required. | |
| With 20 grams of ammonic chloride | 20.0 c.c. |
| " 20 grams of sodium chloride | 20.0 " |
| " 20 grams of sodium acetate | 20.0 " |
| " 0.050 gram of tin, as stannic chloride | 19.6 " |
| Without any addition | 20.0 " |
The interference of the stannic salt is probably mechanical, the precipitate carrying down some arsenious acid.
Effect of Varying Arsenic.—With bulk, starch, and soda as before, but with varying arsenic, the results were:—
| Arsenic added | 1.0 c.c. | 10.0 c.c. | 20.0 c.c. | 50.0 c.c. | 100.0 c.c. |
| "Iodine" required | 1.1 " | 9.9 " | 20.0 " | 50.0 " | 100.0 " |
Determination of Arsenic in Metallic Copper.—Put 1 gram of the copper filings, freed from particles of the file with a magnet, into a 16-oz.-flask; and distil with the ferric chloride mixture, as above described. Neutralise the distillate; acidify; add bicarbonate of soda and starch; dilute; and titrate with the standard solution of iodine.[107] Make a blank determination with 1 gram of electrotype copper, proceeding exactly as with the assay; and deduct the amount of arsenic found in this experiment from that previously obtained.
Working in this way on a copper containing 0.38 per cent. of arsenic and 0.80 per cent. of antimony, 0.38 per cent. of arsenic was found.
Determination of White Arsenic in Crude Arsenic.—Weigh out 1 gram of the dried and powdered substance (or 0.5 gram if rich), and digest with 10 c.c. of a 10 per cent. solution of soda; dilute to about 50 c.c., and filter. Render faintly acid with hydrochloric acid, and filter (if necessary); add 2 or 3 grams of bicarbonate of soda in solution, then 5 c.c. of starch, and titrate the cold solution with the standard solution of iodine.
The following is an example:—
| 1 gram of crude arsenic required 53.7 c.c. "Iodine;" |
| 100 c.c. "Iodine" = 0.6000 gram white arsenic; |
| 100 : 53.7 :: 0.6 : 0.3222, or 32.2 per cent. |
With the test-tube method of dry assaying, this same sample gave results varying from 33 to 35 per cent. of white arsenic, which (judging from its appearance) was impure.[Pg 389]
This may be looked upon as an alternative to the gravimetric method. It is applicable in all cases where the arsenic exists in solution as arsenic acid or as arsenate of soda. The process may be considered in two parts: (1) the preparation of the solution, and (2) the titration.
Preparation of the Solution.—If the arsenic has been separated as sulphide, it is sufficient to attack it with 10 or 15 c.c. of nitric acid, and to heat gently till dissolved, avoiding too high a temperature at first. Afterwards continue the heat till the separated sulphur runs into globules, and the bulk of the acid has been reduced to 3 or 4 c.c. Dilute with 20 or 30 c.c. of water; put in a piece of litmus paper; and add dilute ammonia until just alkaline. Then add 5 c.c. of the sodium acetate and acetic acid solution (which should make the solution distinctly acid); dilute to 150 c.c., and heat to boiling. The solution is ready for titrating.
When the arsenic exists in a nitric acid solution mixed with much copper, it is separated in the way described under Examination of Commercial Copper (Arsenic and Phosphorus), pages 208, 209.
If the arsenic has been separated as ammonium-magnesium arsenate, and phosphates are known to be absent; dissolve the precipitate (after filtering, but without washing) in dilute hydrochloric acid. Add dilute ammonia till a slight precipitate is formed, and then 5 c.c. of the sodium acetate and acetic acid solution; dilute to 150 c.c., and heat to boiling. Titrate.
If phosphates are present (which will always be the case if they were present in the original substance, and no separation with sulphuretted hydrogen has been made), the phosphorus will count in the subsequent titration as arsenic (one part of phosphorus counting as 2.4 parts of arsenic). It will be necessary to dissolve the mixed arsenate and phosphate of magnesia in hydrochloric acid. Add about four or five times as much iron (as ferric chloride) as the combined phosphorus and arsenic present will unite with, and separate by the "basic acetate" process as described under Phosphorus in the Examination of Commercial Copper, page 209. Obviously, when phosphates are present, it is easier to separate the arsenic as sulphide than to precipitate it with the "magnesia mixture."
The Titration.—The standard solution of uranium acetate is made by dissolving 34.1 grams of the salt (with the help of 25 c.c.[Pg 390] of acetic acid) in water; and diluting to 1 litre. The water and acid are added a little at a time, and warmed till solution is effected; then cooled, and diluted to the required volume: 100 c.c. will equal 0.50 gram of arsenic.
The sodic acetate and acetic acid solution is made by dissolving 100 grams of sodic acetate in 500 c.c. of acetic acid, and diluting with water to 1 litre. Five c.c. are used for each assay.
The solution of potassic ferrocyanide used as indicator is made by dissolving 10 grams of the salt in 100 c.c. of water.
To standardise the solution of uranium acetate, weigh up a quantity of white arsenic (As2O3) which shall be about equivalent to the arsenic contained in the assay (0.1 or 0.2 gram); transfer to a flask, and dissolve in 10 c.c. of nitric acid with the aid of heat. Evaporate to a small bulk (taking care to avoid the presence of hydrochloric acid); dilute with water; add a small piece of litmus paper; render faintly alkaline with ammonia; then add 5 c.c. of the sodic acetate mixture; dilute to 150 c.c.; and heat to boiling.
Fill an ordinary burette with the uranium acetate solution, and run into the assay a quantity known to be insufficient. Again heat for a minute or two. Arrange a series of drops of the solution of ferrocyanide of potassium on a porcelain slab, and, with the help of a glass rod, bring a drop of the assay solution in contact with one of these. If no colour is produced, run in the uranium acetate, 1 c.c. at a time, testing after each addition, till a brown colour is developed. It is best to overdo the assay, and to count back. It is not necessary to filter off a portion of the assay before testing with the "ferrocyanide," since the precipitate (uranic arsenate) has no effect.
The following experiments show the effect of variation in the conditions of titration. Make a solution of arsenic acid by dissolving 4.95 grams of arsenious acid (As2O3) in a covered beaker with 35 c.c. of nitric acid; evaporate down to 7 or 8 c.c.; and dilute with water to 1 litre: 100 c.c. will contain 0.375 gram of arsenic. Use 20 c.c. for each experiment.
Effect of Varying Temperature.—It is generally recommended to titrate the boiling solution, since it is possible that the precipitation is only complete on boiling. Low results are obtained in a cold solution, the apparent excess of uranium acetate striking a colour at once; on boiling, however, it ceases to do so; consequently, the solution should always be boiled directly before testing.
In four experiments made in the way described, but with 20 c.c[Pg 391]. of a solution of arsenic acid stronger than that given (100 c.c. = 0.5 gram As), the results at varying temperatures were:—
| Temperature | 15° C. | 30° C. | 70° C. | 100° C. |
| "Uranium" required | 18.0 c.c. | 18.5 c.c. | 18.5 c.c. | 18.7 c.c. |
Effect of Varying Bulk.—These experiments were like those last mentioned, but were titrated boiling, and the volume was varied:—
| Bulk | 50.0 c.c. | 100.0 c.c. | 200.0 c.c. | 300.0 c.c. |
| "Uranium" required | 14.0 " | 14.0 " | 14.5 " | 15.0 " |
Considerable variations in bulk are to be avoided.
Effect of Varying Sodium Acetate.—These experiments were carried out like those last noticed, but the bulk was 150 c.c., and varying amounts of sodic acetate were added in excess of the quantity used in the experiments previously described:—
| Sodic acetate added | 0 gram | 1 gram | 10 grams | 20 grams |
| "Uranium" required | 14.5 c.c. | 14.5 c.c. | 16.0 c.c. | 18.0 c.c. |
It is evidently important that the quantity of this salt present in each titration be measured out, so as to avoid variation.
Effect of Varying the Sodium Acetate and Acetic Acid Solution.—Acetic Acid also affects the results, but in the opposite direction, by preventing the precipitation of uranium arsenate. With varying volumes of the solution now under notice, the results were:—
| Solution added | 0.0 c.c. | 5.0 c.c. | 10.0 c.c. | 15.0 c.c. |
| "Uranium" required | 14.5 " | 14.5 " | 14.5 " | 14.0 " |
| Solution added | 20.0 " | 30.0 " | 40.0 " | 50.0 " |
| "Uranium" required | 13.2 " | 10.0 " | 6.0 " | 2.0 " |
These show that the quantity ordered (5 c.c.) must be adhered to.
Effect of Foreign Salts.—In these experiments, 10 grams of the salt (the effect of which it was desired to determine) were added to a solution in other respects resembling those previously used:—
| Salt added | Ammonic sulphate | Ammonic nitrate | Ammonic chloride | Magnesium sulphate |
| "Uranium" required | 15.5 c.c. | 15.5 c.c. | 15.3 c.c. | 15.3 c.c. |
Without any addition, 15.0 c.c. were required; and in another experiment, in which 30 grams of ammonic salts were present,[Pg 392] 15.6 c.c. of uranium solution were required. Such variations in the amount of ammonic salts as occur in ordinary working are unimportant.
Phosphates, of course, interfere. In fact, the uranium acetate solution can be standardised by titrating with a known weight of phosphate, and calculating its equivalent of arsenic. Thus, in an experiment with 0.6 gram of hydric sodic phosphate (Na2HPO4.12H2O), equivalent to 0.05195 gram of phosphorus, or 0.1256 gram of arsenic, 23.25 c.c. of a solution of uranium acetate were required. The same solution standardised with white arsenic gave a standard of which 100 c.c. = 0.5333 gram arsenic. On this standard the 0.6 gram of sodic phosphate should have required 23.5 c.c.
Experiments in which 0.1 gram of bismuth and 0.1 gram of antimony were present with 0.1 gram of arsenic, showed no interference on the titration. Ferric or aluminic salts would remove their equivalent of arsenic, and, consequently, must be removed before titrating.
Effect of Varying Arsenic.—Varying amounts of metallic arsenic were weighed up and dissolved in nitric acid, &c., and titrated:—
| Arsenic taken | 0.010 gram | 0.050 gram | 0.100 gram | 0.200 gram |
| Arsenic found | 0.010 " | 0.050 " | 0.100 " | 0.197 " |
These experiments show that the method yields good results within these limits.
Determination of Arsenic in Mispickel.—Weigh up 1 gram of the dried and powdered ore, and evaporate to near dryness with 20 c.c. of dilute nitric acid. Make up to 100 c.c. with water, and pass sulphuretted hydrogen to reduce the ferric iron to the ferrous state, then add 20 c.c. of dilute ammonia, and again pass sulphuretted hydrogen. Warm, filter, and evaporate the filtrate to drive off the excess of ammonia; then add 10 c.c. of nitric acid, and boil down till the sulphide of arsenic at first precipitated is dissolved; neutralise; add 5 c.c. of sodium acetate and acetic acid solution; transfer to a pint flask, boil, and titrate.
For example, an impure sample of ore required, in duplicate assay of half a gram each, when treated in the above-mentioned way, 39.6 and 39.5 c.c. of the uranium acetate solution (100 c.c. = 0.537 gram of arsenic), equivalent to 0.2114 gram of arsenic, or 42.3 per cent.
An alternative method is as follows. Powder the ore very finely and weigh up .5 gram. Place in a 2-3/4 inch berlin dish and add strong nitric acid, one drop at a time until the action ceases; with care there need be no very violent reaction. Dry over a[Pg 393] water bath. Cover with 2 grams of nitre and over this spread 5 grams of a mixture of equal parts of nitre and carbonate of soda. Fuse in a muffle or over a large gentle blow-pipe flame for 4 or 5 minutes. This will spoil the dish. Allow to cool and boil out in a larger dish with 100 c.c. of water. Filter and wash into an 8 oz. flask. Acidify the liquor with nitric and boil down to about 100 c.c. The acid should not be in too large excess, but an excess is needed to destroy nitrites. Neutralise with soda or ammonia. Add 5 c.c. of the mixture of sodium acetate and acetic acid. Titrate with uranium acetate.
Determination of Arsenic (As) in Crude Arsenic.—The method given under the iodine titration simply determines that portion of the arsenic which is present in the substance as arsenious oxide or white arsenic. The following method will give the total arsenic in the sample. It would be incorrect to report this as so much per cent. of arsenious oxide, although it may be reported as so much per cent. of arsenic equivalent to so much per cent. of white arsenic, thus:—
| Arsenic | 30.0 per cent. |
| Equivalent to white arsenic | 39.6 " |
The equivalent of white arsenic is calculated by multiplying the percentage of arsenic by 1.32. The method of determining the percentage of arsenic is as follows:—-Boil 1 gram of the sample with 10 c.c. of nitric acid. When the bulk of the solution has been reduced to one-half, and red fumes are no longer evolved, dilute with a little water, and filter into a flask. Neutralise the filtrate, add 5 c.c. of sodic acetate solution, boil and filter. The precipitate (ferric arsenate) is transferred to a small beaker, treated with 5 c.c. of dilute ammonia, and sulphuretted hydrogen passed through it. The iron sulphide is filtered off, and the filtrate evaporated with an excess of nitric acid. When the solution is clear, it is neutralised, and 1 or 2 c.c. of sodic acetate solution having been added, is then mixed with the first filtrate. The solution is boiled and titrated.
A sample treated in this way required 49.2 c.c. of the uranium acetate solution (100 c.c. = 0.537 gram of arsenic), equivalent to 26.4 per cent.
Determination of Arsenic in Brimstone.—Take 10 grams of the substance, and powder in a mortar; rub up with 10 c.c. of dilute ammonia and a little water; rinse into a pint flask; pass a current of sulphuretted hydrogen; and warm on a hot plate for a few minutes. Filter, acidulate the filtrate with sulphuric acid; filter off the precipitate; attack it with 10 c.c. of nitric acid; and proceed as in the other determinations.[Pg 394]
1. Mispickel contains 45.0 per cent. of arsenic, to how much white arsenic will this be equivalent?
2. How would you make a standard solution of iodine so that 100 c.c. shall be equivalent to 1 gram of white arsenic?
3. What weight of arsenic is contained in 1 gram of pyrarsenate of magnesia, and what weight of ammonic-magnesic arsenate would it be equivalent to?
4. The residue, after heating 10 grams of crude arsenic, weighed 0.62 gram. What information does this give as to the composition of the substance? If another 10 grams of the substance, heated on a water-bath, lost 0.43 gram, what conclusions would you draw, and how would you report your results?
5. If a sample of copper contained 0.5 per cent. of arsenic, and 1 gram of it were taken for an assay, how much standard uranium acetate solution would be required in the titration?
Phosphorus rarely occurs among minerals except in its highest oxidized state, phosphoric oxide (P2O5), in which it occurs abundantly as "rock phosphate," a variety of apatite which is mainly phosphate of lime. Phosphates of most of the metallic oxides are found. Phosphoric oxide in small quantities is widely diffused, and is a constituent of most rocks. Its presence in varying amounts in iron ores is a matter of importance, since it affects the quality of the iron obtainable from them.
Phosphorus occurs in alloys in the unoxidized state. It is directly combined with the metal, forming a phosphide. In this manner it occurs in meteoric iron. The alloy phosphor-bronze is made up of copper, tin, zinc, and phosphorus.
Phosphates are mined in large quantities for the use of manure manufacturers, and for making phosphorus.
Phosphorus and arsenic closely resemble each other in their chemical properties, more especially those which the assayer makes use of for their determination. Phosphorus forms several series of salts; but the phosphates are the only ones which need be considered. Pyrophosphate of magnesia, which is the form in which phosphoric oxide is generally weighed, differs from the ordinary phosphate in the proportion of base to acid. Metaphosphates differ in the same way. If these are present, it must be remembered they act differently with some reagents from the ordinary phosphates, which are called orthophosphates. They are, however, all convertible into orthophosphates by some means[Pg 395] which will remove their base, such as fusion with alkaline carbonates, boiling with strong acids, &c.[108]
Phosphides are converted into phosphates by the action of nitric acid or other oxidizing agents. Dilute acids, when they act on the substance, evolve phosphuretted hydrogen (PH3). The student should be on his guard against losing phosphorus in this manner.
There is no dry assay for phosphorus. All assays for it are made either gravimetrically or volumetrically.
The separation of phosphoric oxide is made as follows:—The ore or metal is dissolved in acid and evaporated, to render the silica insoluble. It is taken up with hydrochloric acid, diluted with water, and treated with sulphuretted hydrogen. The filtrate is boiled, to get rid of the excess of gas, and treated with nitric acid, to peroxidize the iron present. If the iron is not present in more than sufficient quantity to form ferric phosphate with all the phosphorus present, some ferric chloride is added. The iron is then separated as basic acetate. The precipitate will contain the phosphorus, together with any arsenic acid not reduced by the sulphuretted hydrogen. The precipitate should have a decided brown colour. The precipitate is washed, transferred to a flask, and treated first with ammonia, and then with a current of sulphuretted hydrogen. The filtrate from this (acidulated with hydrochloric acid, and, if necessary, filtered) contains the phosphorus as phosphoric acid. This method is not applicable in the presence of alumina, chromium, titanium, or tin, if the solution is effected with nitric acid. The precipitate obtained by the action of nitric acid on tin retains any phosphoric or arsenic oxide that may be present.
A method of separation more generally applicable and more convenient to work is based on the precipitation of a yellow phospho-molybdate of ammonia,[109] by the action of an excess of ammonic molybdate upon a solution of a phosphate in nitric acid. Dissolve the substance by treatment with acid, and evaporate to dryness. Take up with 10 c.c. of nitric acid, and add 20 grams of ammonic nitrate, together with a little water. Next put in the solution of ammonium molybdate solution in the proportion of about 50 c.c. for each 0.1 gram of phosphoric oxide judged to be present. Warm to about 80° C., and allow to stand for an hour. Filter, and wash[Pg 396] with a 10 per cent. solution of ammonic nitrate. It is not necessary that the whole of the precipitate be placed on the filter; but the beaker must be completely cleaned. Dissolve the precipitate off the filter with dilute ammonia, and run the solution into the original beaker. Run in from a burette, slowly and with stirring, "magnesia mixture," using about 15 c.c. for each 0.1 gram of phosphoric oxide. Allow to stand for one hour. The white crystalline precipitate contains the phosphorus as ammonium-magnesium phosphate.
Phosphate of lead is decomposed by sulphuric acid; the lead is converted into the insoluble lead sulphate, and the phosphoric acid is dissolved. Phosphate of copper and phosphate of iron may be treated with sulphuretted hydrogen; the former in an acid, and the latter in an alkaline, solution. Phosphate of alumina is generally weighed without separation of the alumina, since this requires a fusion. In all cases the aim is to get the phosphoric oxide either free, or combined with some metal whose phosphate is soluble in ammonia.
Joulie's method of separation is as follows:—One to ten grams of the sample are treated with hydrochloric acid, and evaporated to dryness with the addition (if any pyrites is present) of a little nitric acid. The residue is taken up with hydrochloric acid, cooled, transferred to a graduated flask, and diluted to the mark. It is then shaken up, filtered through a dry filter, and a measured portion (containing about 0.05 gram of phosphoric acid) transferred to a small beaker. Ten c.c. of a citric-acid solution of magnesia[110] is added, and then an excess of ammonia. If an immediate precipitate is formed, a fresh portion must be measured out and treated with 20 c.c. of the citrate of magnesia solution and with ammonia as before. The beaker is put aside for from two to twelve hours. The precipitate is then filtered off and washed with weak ammonia; it contains the phosphorus as ammonium-magnesium phosphate.
If the phosphate is not already in the form of ammonic-magnesic phosphate, it is converted into this by the addition to its solution of an excess of ammonia and "magnesia mixture." In order to get the precipitate pure, the "magnesia mixture" is run in[Pg 397] gradually (by drops) from a burette, with constant stirring. A white crystalline precipitate at once falls, if much phosphorus is present; but, if there is only a small quantity, it may be an hour or two before it shows itself. The solution is best allowed to rest for twelve or fifteen hours (overnight) before filtering. The presence of tartaric acid should be avoided; and the appearance of the precipitate should be crystalline. The solution is decanted through a filter, and the precipitate washed with dilute ammonia, using as little as may be necessary. The precipitate is dried, transferred to a weighed Berlin or platinum crucible; the filter-paper is carefully burnt, and its ash added to the precipitate, which is then ignited, at first gently over a Bunsen burner, and then more strongly over the blowpipe or in the muffle. The residue is a white mass of magnesium pyrophosphate containing 27.92 per cent. of phosphorus, or 63.96 per cent. of phosphoric oxide.
Instead of separating and weighing this compound, the phosphoric oxide in it can be determined by titration. In many cases the ore may be dissolved and immediately titrated without previous separation. It is better, however, to carry the separation so far as to get phosphoric acid, an alkaline phosphate, or the magnesia precipitate. It may then be prepared for titration in the following way:—The precipitate in the last case (without much washing) is dissolved in a little hydrochloric acid, and the solution in any case rendered fairly acid. Dilute ammonia is added till it is just alkaline, and then 5 c.c. of the sodic acetate and acetic acid mixture (as described under the Arsenic Assay). This should yield a clear distinctly-acid solution. It is diluted to 100 or 150 c.c., heated to boiling, and titrated with the uranium acetate solution, using that of potassic ferrocyanide as indicator.
The standard solution required is made by dissolving 35 grams of uranium acetate in water with the aid of 25 c.c. of acetic acid, and diluting to 1 litre.
An equivalent solution of phosphoric oxide is made by dissolving 25.21 grams of crystallised hydric disodic phosphate (HNa2PO4.12H2O) in water, and making up to 1 litre. 100 c.c. will contain 0.5 gram of phosphoric oxide (P2O5), or 0.2183 gram of phosphorus. In making this solution, transparent crystals only must be used. The uranium acetate solution is only approximately equivalent to this, so that its exact standard must be determined.[Pg 398]
Sodic Acetate and Acetic Acid Solution.—It is the same as that described under Arsenic.[111] Use 5 c.c. for each assay.
The following experiments show the effect of variation in the conditions of the titration:—
Effect of Varying Temperature.—The solution should be titrated while boiling. This is especially necessary for the last few c.c. in order to get a decided and fixed finishing point.
| Temperature | 15° C. | 30° C. | 70° C. | 100° C. |
| "Uranium" required | 18.0 c.c. | 19.2 c.c. | 19.0 c.c. | 18.9 c.c. |
Effect of Varying Bulk.—
| Bulk | 50.0 c.c. | 100.0 c.c. | 200.0 c.c. | 300.0 c.c. |
| "Uranium" required | 18.8 " | 18.9 " | 19.0 " | 19.3 " |
Variation in bulk affects the results; therefore, a constant bulk should be adhered to.
Effect of Varying Sodium Acetate and Acetic Acid Solution.—
| Sodium acetate and acetic acid solution | 0.0 c.c. | 1.0 c.c. | 5.0 c.c. | 10.0 c.c. | 20.0 c.c. |
| "Uranium" required | 18.9 " | 18.9 " | 19.0 " | 18.8 " | 17.5 " |
As in the titration with arsenates, an excess is dangerous to the assay; a definite quantity (5 c.c.) should, therefore, be used.
Effect of Foreign Salts.—Besides the sodium acetate, &c., added, the only salts likely to be present are those of ammonia and magnesia. In three experiments, in one of which no foreign salts were introduced, while in the other two 5 grams of ammonic chloride and of magnesium sulphate respectively were added, there were required:—
| With ammonic chloride | 18.8 c.c. | "Uranium" solution |
| With magnesium sulphate | 19.0 " | " |
| Without foreign salts | 18.9 " | " |
Effect of Varying Phosphate.—
| "Phosphate" solution added | 10.0 c.c. | 20.0 c.c. | 50.0 c.c. | 100.0 c.c. |
| "Uranium" required | 9.8 " | 18.9 " | 47.6 " | 94.5 " |
The quantity of phosphoric oxide in the assay solution for the conditions of titration should not be much less than 0.05 gram.[Pg 399] For smaller quantities the uranium solution should be diluted to half its strength, and the assay solution concentrated by reducing its bulk to 50 c.c. and using 2.5 c.c. of the sodium acetate and acetic acid solution.
Determination of Phosphoric Oxide in Apatite.—Weigh up 0.5 gram of the dried and powdered sample, and dissolve it in 5 c.c. of hydrochloric acid. Evaporate to a paste, add 5 c.c. of the sodic acetate and acetic acid solution, dilute to 100 c.c. with water, boil, and titrate with uranium acetate solution.
In an example, 0.5 gram of apatite required 37.4 c.c. of uranium acetate solution (standard equal to 0.5291 gram of phosphoric oxide). The sample therefore contained 0.1979 gram of P2O5, equal to 39.58 per cent.
Determination of Phosphoric Oxide in an Iron Ore.—Take 10 grams, boil with 50 c.c. of hydrochloric acid, and evaporate to a paste; take up with 10 c.c. of dilute hydrochloric acid, and dilute with water to 400 c.c. Pass sulphuretted hydrogen for nearly a quarter of an hour; warm, and filter. Boil off the excess of gas; cool, add ammonia till nearly neutral, and then a few drops of ferric chloride solution, and 4 or 5 grams of sodium acetate, with a drop or two of acetic acid. Boil and filter. Dissolve the precipitate in hot dilute hydrochloric acid, and add citro-magnesia mixture and ammonia; allow to stand overnight; filter, ignite, and weigh.
In an example, 10 grams of ore gave 28.5 milligrams of magnesic pyrophosphate, which is equivalent to 0.18 per cent. of phosphoric oxide.
Determination of Phosphorus in Iron.—Take from 2 to 10 grams (according to the amount of phosphorus present), and dissolve in aqua regia, keeping the nitric acid in excess; evaporate to dryness and take up with hydrochloric acid, boil, dilute, and filter. Add 10 c.c. of nitric acid, nearly neutralise with ammonia, render acid with 3 or 4 c.c. of nitric acid, and add 10 or 20 c.c. of ammonic molybdate solution. Heat for some time, allow to settle, filter, and wash the precipitate with a solution of ammonic nitrate. Dissolve the precipitate in dilute ammonia, nearly neutralise with dilute hydrochloric acid, and add first "magnesia mixture," and then ammonia; allow to stand overnight; filter, wash with dilute ammonia, dry, ignite, and weigh as magnesic pyrophosphate. Calculate to phosphorus.[Pg 400]
1. Ten grams of an iron yielded 12 milligrams of pyrophosphate of magnesia. What percentage of phosphorus did the metal contain?
2. Ten grams of an iron ore gave 12 milligrams of pyrophosphate. What percentage of phosphoric oxide did it contain?
3. What weight of apatite 3Ca3(PO4)2.CaClF would require 50 c.c. of standard uranium solution (100 c.c. equal to 0.5 gram of P2O5)?
4. You have reason to believe that a precipitate which has been weighed as magnetic pyrophosphate contains some arsenate. How would you determine the amount of phosphate really present?
5. Twenty c.c. of a solution of sodic phosphate containing 0.100 gram of P2O5 was found to require a solution containing 0.700 gram of hydrated uranium acetate in a titration. The precipitate contains 80.09 per cent. uranium oxide and 19.91 per cent. of phosphoric oxide. What percentage of uranium oxide was contained in the uranic acetate?
Nitrogen occurs in nature in the free state, and forms about four-fifths of the atmosphere. In combination, as nitrate, it is found in nitre (KNO3), and Chili saltpetre (NaNO3), minerals which have a commercial importance. The latter occurs in beds, and is extensively worked for use as a manure and in the preparation of nitric acid.
Nitrogen is mainly characterised by negative properties, although many of its compounds are very energetic bodies. It is a gas, present everywhere, but so inactive that the assayer can always afford to ignore its presence, and, except in testing furnace gases, &c., he is never called on to determine its quantity.
The nitrates are an important class of salts, and may be looked on as compounds of the bases with nitric pentoxide (N2O5). They are, with the exception of a few basic compounds, soluble in water, and are remarkable for the ease with which they give up their oxygen. The alkaline nitrates fuse readily, and lose oxygen with effervescence forming nitrites; while at a higher temperature they yield more oxygen and lose their nitrogen, either as a lower oxide or as nitrogen. The nitrates of the metals, on heating, leave the oxide of the metal. It is as yielders of oxygen that nitrates are so largely used in the manufacture of explosives. Gunpowder contains from 65 to 75 per cent. of potassium nitrate (nitre).
Nitrates are best detected and determined by their yielding nitric oxide when treated with sulphuric acid and a suitable reducing agent, such as ferrous sulphate, mercury, or copper. Nitric oxide is a colourless gas very slightly soluble in water. It[Pg 401] combines at once with oxygen, on mixing with the air, to form brown "nitrous fumes," and dissolves in a solution of ferrous sulphate, producing a characteristic blackish-brown colour. It is this colour which affords the best and most easily-applied test for nitrates. The substance suspected to contain nitrates is dissolved in about 1 c.c. of water, and treated with an equal volume of strong sulphuric acid. After cooling, a solution of ferrous sulphate is poured on its surface, so as to form a layer resting on it. On standing, a brown or black ring is developed where the liquids join, if any nitrate or nitrite is present. Nitrites are distinguished from nitrates by effervescing and yielding brown fumes when treated with a little dilute sulphuric acid.
The separation of nitrates is in many cases difficult. Generally, on treating the substance with water, the nitrate will be in the solution, and is filtered off from any insoluble matter. In the exceptional cases it is got into solution by treating with a boiling solution of sodium carbonate; the nitrate will contain it as an alkaline nitrate.
Since, however, in their determination, nitrates are never separated and weighed as such, the difficulty of separating them has little importance. Usually, the determination can be made on the original aqueous solution, and it is never necessary to do more than remove any special substance which has a bad effect; and this is easily done by the usual reagents.
It follows from what has been said that there is no direct gravimetric determination. The percentage of nitrogen pentoxide (N2O5) in a comparatively pure nitrate is sometimes determined indirectly in the following way:—Place in a platinum-crucible 4 or 5 grams of powdered and cleaned quartz. Ignite, cool in a desiccator, and weigh with the cover. Mix 1 gram of the dried and powdered salt with the quartz in the crucible by stirring with a stout platinum-wire. Cover the crucible, and heat in a Bunsen-burner flame at scarcely visible redness for half-an-hour. Cool and weigh. The loss in weight gives the amount of nitrogen pentoxide. Sulphates and chlorides in moderate quantity do not interfere. The following is an example of the process:[Pg 402]—
| Crucible and sand | 26.6485 grams |
| Nitre taken | 1.0000 " |
| ———— | |
| 27.6485 " | |
| Weight after ignition | 27.1160 " |
| ———— | |
| Loss on ignition | 0.5325 " |
This is equal to 53.25 per cent. of nitrogen pentoxide.
This is based on the oxidising action of nitric acid, or of nitrates in acid solutions on ferrous salts. The pentoxide (N2O5) of the nitrate is reduced to nitric oxide (NO), so that 336 parts of iron peroxidised represent 108 parts of nitric pentoxide as oxidising agent.[112] The quantity of iron peroxidised is determined by taking a known quantity of ferrous salt, oxidizing with a weighed sample of nitrate, and then determining the residual ferrous iron by titration with bichromate or permanganate of potassium solution. The difference between the ferrous iron taken and that found, gives the amount oxidized by the nitrate. The speed with which nitric oxide takes up oxygen from the air, and thus becomes capable of oxidising more iron, renders some precautions necessary; ferrous chloride should, therefore, be used, since it is easier to expel nitric oxide (by boiling) from solutions of a chloride than it is from those of a sulphate. The process is as follows:—Dissolve 2 grams of thin soft iron wire in 50 c.c. of hydrochloric acid in a flask provided with an arrangement for maintaining an atmosphere of carbon dioxide. When the iron has dissolved, allow the solution to cool, and add 0.5 gram of the nitrate. Heat gently for a few minutes, and then boil until the nitric oxide is expelled. An atmosphere of carbon dioxide must be kept up. Dilute with water, and titrate the residual iron with standard solution of bichromate of potassium. The standard "bichromate" is made by dissolving 17.5 grams of the salt (K2Cr2O7) in water, and diluting to 1 litre: 100 c.c. equal 2 grams of iron. Deduct the weight of iron found from the 2 grams originally taken, and multiply by 0.3214. This gives the weight of the pentoxide in the sample. In an example, 0.5 gram of nitre was taken, and 59.4 c.c. of the "bichromate" solution were required. The 59.4 c.c. thus used are equivalent to 1.198 gram of iron. This leaves 0.822 gram as the quantity oxidised by the nitre, which, multiplied by 0.3214, gives 0.2642 gram for the nitrogen pentoxide, or 52.8 per cent.[Pg 403]
This is based upon the measurement of the nitric oxide evolved on shaking up a weighed quantity of the nitrate with sulphuric acid over mercury in a nitrometer. Each c.c. of nitric oxide obtained, when reduced to normal temperature and pressure, is equivalent to:—
| 0.627 | milligram | of nitrogen. |
| 1.343 | " | of nitric oxide. |
| 2.418 | " | of nitric pentoxide. |
| 2.820 | " | of nitric acid. |
| 3.805 | " | of sodium nitrate. |
| 4.523 | " | of potassium nitrate. |
In working on substances not rich in nitrates, an ordinary nitrometer (fig. 69) is used; but in the assay of sodium nitrate, nitroglycerine, &c., an instrument provided with a bulb having a capacity of 100 c.c. is employed.
The plan of working is as follows:—The "measuring tube" is filled with mercury until it reaches up into the tap, and the levelling-tube is placed so that it contains an inch or two of mercury. If the nitrate is in solution, 2 or 3 c.c. of the liquid (dilute liquids are brought to this bulk by evaporation) are measured into the cup. The levelling-tube is lowered a little, and the tap cautiously opened until all but the last drop of the liquid has run in. The cup is then rinsed with 2 or 3 c.c. of sulphuric acid, which is run in in the same way, and the operation is repeated with another lot of acid. The measuring-tube is now taken from the clamp, and shaken for two or three minutes, until no more gas is given off. It is replaced, and the mercury-level in the two tubes adjusted. Then it is allowed to stand until the froth has subsided, and the gas has cooled to the temperature of the room. The volume of the gas is then read off. In adjusting the level, account must be taken of the sulphuric acid in the measuring-tube; this is allowed for by having the mercury higher in the other tube by,[Pg 404] say, 1 mm. for each 6.5 mm. of sulphuric acid, or it is counterpoised by an equal height of sulphuric acid in the levelling-tube, in which case the two mercury-levels are made to correspond. On opening the tap after reading off the volume, there should be no change in the level of the mercury. If it should rise or fall a little, a slight increase or decrease (say 0.1 c.c.) is made to the volume previously read off.
In working with nitrate of soda, &c., in the bulb nitrometer, it is necessary to take a quantity of the substance which will yield more than 100 and less than 150 c.c. of the gas.
[103] Na3AsO3 + H2O + 2I = Na3AsO4 + 2HI. The acid is at once neutralised.
[104] Mr. Thomas Gibb is the originator of this ingenious process.
[105] By taking hold of the water present, it may prevent the dissociation of arsenious chloride.
[106] It is difficult to get ferric chloride free from arsenic; but the following treatment will remove 80 or 90 per cent. of the arsenic contained in the commercial material:—Dissolve 2 or 3 lbs. of ferric chloride with the smallest amount of water that will effect solution with the addition of 100 c.c. of hydrochloric acid; add a solution of sulphurous acid in quantity sufficient to reduce 2 or 3 per cent. of the iron to the ferrous state; allow to stand a week; and then boil, to remove the hydrochloric acid added. Nitric acid, which is prejudicial, is also removed by this treatment.
[107] When the amount of arsenic to be estimated is small (as in refined coppers), it is better to use a weaker solution of iodine. This is made by diluting 200 c.c. of the standard solution with water to 1 litre. Each c.c. will equal 0.1 per cent., if 1 gram of the metal has been taken for the assay.
[108] The constitution of these phosphates may be thus
illustrated—
Magnesic meta-phosphate MgO.P2O5.
Magnesic pyro-phosphate 2MgO.P2O5.
Magnesic ortho-phosphate 3MgO.P2O5.
[109] The composition of which is—
MoO2 90.74,
P2O5 3.14,
(NH4)2O 3.57,
H2O 2.55=100.00.
[110] This is made by adding 27 grams of magnesium carbonate (a little at a time) to a solution of 270 grams of citric acid in 350 c.c. of warm water; and, when dissolved, adding 400 c.c. of dilute ammonia, and making up the bulk to 1 litre; 20 c.c. of the solution is sufficient for 0.1 gram of P2O5, although more will be required if much iron or alumina is present.
[111] For the details of the titration, the student is referred to the same place.
[112] N2O5 + 6FeO = 3Fe2O3 + 2NO.
In assaying, more especially products direct from the mine, there is always found, when the rock is siliceous, a quantity of white sandy-looking substance, insoluble in acids, which is sometimes accompanied by a light gelatinous material very difficult to filter. This is variously described as "insoluble," "sand," "insoluble silicates," "gangue," or "rocky matter." It may be pure quartz; but oftener it is mixed with silicates from the rock containing the mineral. Some silicates, but not many, are completely decomposed by boiling with hydrochloric acid or aqua regia; and others are partly so, they yield a gelatinous precipitate of silica which greatly interferes with the filtering. It is a common practice with assayers to carry the first attack of the sample with acids to dryness, and to take up with a fresh portion of acid. By this means the separated silica becomes granular and insoluble, and capable of being filtered off and washed with comparative ease.
This residue may be ignited and weighed; and be reported as so much per cent. of "silica and silicates insoluble in acids." Unless specially wanted, a determination of its constituents need not be made. When required, the analysis is best made on the ignited residue, and separately reported as "analysis of the insoluble portion."
Silicon only occurs in nature in the oxidised state; but the oxide generally known as silica (SiO2) is common, being represented by the abundant minerals—quartz, flint, &c. Silica, combined with alumina, lime, oxide of iron, magnesia and the alkalies, forms a large number of rock-forming minerals. Most rock masses, other than limestones, contain over 50 per cent. of silica. The following are analyses of some of the commoner silicates; but it must be noted that these minerals often show great variation in composition. This is more especially true of chlorite, schorl, hornblende and augite.[Pg 406]
| Silica, SiO2. | Alumina, Al2O3. | Ferric Oxide, Fe2O3. | Ferrous Oxide, FeO. | Lime, CaO. | Magnesia, MgO. | Potash, K2O. | Soda, Na2O. | Fluorine, Water &c. | |
| Potash-felspar | 65.2 | 18.2 | 0.2 | — | — | — | 14.7 | 1.5 | |
| Soda-felspar | 67.0 | 19.2 | — | 0.3 | 1.2 | 1.8 | 2.2 | 7.2 | |
| Lime-felspar | 43.3 | 35.4 | — | 1.3 | 17.4 | 0.35 | 0.5 | 0.9 | |
| Potash-mica | 45.7 | 33.7 | 3.1 | — | — | 1.1 | 7.5 | 2.8 | F (0.8) H2O (4.9) |
| Magnesia-mica | 39.1 | 15.4 | 7.1 | — | — | 23.6 | 7.5 | 2.6 | F (0.7) |
| Hornblende | 40.6 | 14.3 | 5.8 | 7.2 | 12.5 | 14.0 | 1.5 | 1.6 | |
| Augite | 50.0 | 3.7 | 2.4 | 6.6 | 22.8 | 13.5 | — | — | MnO (0.1) |
| Almandine (Garnet) | 39.7 | 19.7 | — | 39.7 | — | — | — | — | MnO (1.8) |
| Chlorite (Peach) | 32.1 | 18.5 | — | — | — | 36.7 | — | — | H2O (12.1) |
| Schorl | 37.0 | 33.1 | 9.3 | 6.2 | 0.5 | 2.6 | 0.7 | 1.4 | B2O3 (7.7) F (1.5) |
| China-clay | 46.7 | 39.6 | — | — | — | — | — | — | H2O (13.4) |
| Talc | 61.7 | — | — | 1.7 | — | 31.7 | — | — | H2O (3.8) |
| Serpentine | 42.9 | — | — | 3.8 | — | 40.5 | — | — | H2O (12.6) |
| Olivine | 39.3 | — | — | 14.8 | — | 45.8 | — | — |
Silicon, from a chemical point of view, is an interesting body. It combines with iron to form a silicide; and is present in this condition in cast iron. Only in the case of the analysis of this and similar substances is the assayer called on to report the percentage of silicon. Silicon is readily converted into silica by the action of oxidizing agents. Silica forms only one series of salts—the silicates—which have in many cases a complex constitution; thus there are a large number of double silicates, which vary among themselves, not only in the relation of base to acid (which is the essential difference), but also in the ratio of the bases between themselves (which varies with almost every specimen).
Silica is detected by heating the substance with a fluoride and sulphuric acid in a platinum-crucible. On holding a rod, moistened with a drop of water, over the escaping fumes, the white crust of silica formed on the drop of water shows its presence. The insolubility of a fragment of the mineral in a bead of microcosmic salt, is also a very good test; the fragment, on prolonged heating, does not lose its angular form.
There is no dry assay for this substance, nor volumetric method; when the determination is required, it is carried out gravimetrically and, generally, by the following plan.
If the sample contains oxides, sulphides, &c., in any quantity, these are first dissolved out by treatment with acid, evaporated to dryness, taken up with hydrochloric acid, and filtered. The dried residue is treated in the same way as the silicates. Some silicates are completely decomposed by such treatment; but it saves time (unless one is sure that no undecomposable silicate is present) to treat these in the same way as the others. On the other hand, there are some silicates which are only attacked with difficulty even by fusion with alkaline carbonates; consequently, it is always well to have the substance reduced to the finest state of division by careful powdering, as this greatly assists the subsequent action. With very hard silicates, the grinding away of the mortar in this operation will be perceptible; the foreign matter thus introduced must not be ignored. Previously igniting the substance sometimes assists the powdering; but it is best to use a steel mortar. The particles of steel can be removed by a magnet, or, where the nature of the substance will allow it, by boiling with a little dilute hydrochloric acid.
The dried and powdered material is intimately mixed with four times its weight of "fusion mixture" in a platinum-crucible or dish. It is then moderately heated over a Bunsen burner, and afterwards more strongly fused over a blast, or enclosed in a clay crucible in the wind-furnace. The action is continued until the fused mass is perfectly tranquil. With very refractory substances,[Pg 408] the action must be long continued at a high temperature. When sufficiently cold, the crucible is examined to see that no particles of foreign matter are adhering to its outer surface. It is then transferred to a five- or six-inch evaporating-dish, where its contents are acted upon with warm water for some time. The "melt" will slowly dissolve, but the solution should be hastened by keeping the liquid moderately acid with hydrochloric acid. When the "melt" has dissolved, clean and remove the platinum-dish, and evaporate the solution to a paste. Continue the evaporation to dryness on a water-bath (not on the hot plate), and whilst drying stir with a glass rod, feeling at the bottom of the dish for any unfused particles, which, if present, can be detected by their grittiness. If there is much grit, it will be necessary to repeat the assay; but with a small quantity it will only be necessary to refuse the grit and silica after ignition.
During solution of the "melt" and evaporation (which may be carried on together), a clear solution will not be obtained, a flocculent silica will separate out, and towards the end of the evaporation the mass will get gelatinous. The drying of the jelly must be finished on the water-bath; first, because at this temperature the silica is rendered insoluble in hydrochloric acid, whilst the solubility of the alumina, iron, &c., is unaffected, which would not be the case at a much higher temperature; and second, because the gelatinous residue requires very cautious drying to prevent loss from spirting.
When dry, the substance is moistened, and heated with strong hydrochloric acid, and the sides of the dish are washed down with water. The silica is washed by decantation two or three times with hydrochloric acid and hot water, before being thrown on to the filter. The filtrate is again evaporated to dryness, taken up with a little hydrochloric acid and water and again filtered. The residue on the filter is silica. The two lots of silica are washed free from chlorides with hot water, dried on an air-bath, transferred to a platinum-crucible, ignited gently at first, at last strongly over the blast or in a muffle, cooled in a desiccator, and weighed.
The white powdery precipitate is silica (SiO2), and its weight, multiplied by 100, and divided by the weight of ore taken, gives the percentage of silica in the sample. Where the percentage of silicon is wanted, which is very rarely the case, it is got by multiplying this result by 0.4667. It is always necessary to examine the purity of the body weighed as silica. This is done by re-fusing the material weighed, and re-determining the silica in it; or, better, by mixing a weighed portion in a platinum-dish with a little strong sulphuric acid, covering with hydrofluoric acid, and[Pg 409] evaporating. In the latter case, the silica will be converted into fluoride, which will be driven off, and the impurities will be left behind as sulphates of barium, phosphate and oxide of tin, titanium, &c. This must be weighed and deducted from the weight of the silica. In a complete examination of a silicate it should be treated with the precipitate containing alumina, ferric oxide, &c.
The student interested in the analysis of rocks and rock-forming minerals is advised to consult a valuable paper by Dr. W.F. Hillebrand in the Bulletin of the United States Geological Survey, No. 148, to which I am very largely indebted in the revision of the following pages.
Moisture.—Five grams of the powdered sample is dried between watch-glasses in the water-oven for two hours, or till its weight is constant; and the loss is reported as water lost at 100° C. The rest of the determinations are made on this dried mineral.
Combined Water, &c.—Weigh up 1 gram of the substance, and ignite over the blowpipe for some time in a platinum-crucible, cool in a desiccator, and weigh. Record the loss as "loss on ignition," not as "combined water."
Silica.—The ignition should have been performed in an oxidising atmosphere in a muffle or over a slanting blowpipe flame; this will ensure the oxidation of any pyrites or other sulphide present, which if unoxidised would injure the crucible in the next operation. The ignited residue is mixed with 6 or 7 grams of anhydrous sodium carbonate. This reagent should be the purest obtainable, but its purity should be checked, or rather its impurities should be determined by running a "check" or "blank" assay with 10 grams of it through the stages of the analysis; the impurities will be chiefly silica, alumina and lime, and altogether they ought not to exceed 1 milligram. The crucible with the mixture is heated at first gently over a Bunsen and afterwards more strongly in an oxidising atmosphere in a muffle or over the blowpipe. The fused mass is allowed to cool in the crucible, and is then dissolved out in a basin with water and a small excess of hydrochloric acid. After the removal and cleaning of the crucible, the liquor is evaporated almost to dryness. Dr. Hillebrand advises stopping short of complete dryness. The residue is taken up with a little hydrochloric acid and water and filtered and washed. The liquor, including the washings, is again[Pg 410] evaporated and taken up with water and a little acid. Usually about 1 per cent. of silica will be thus recovered. It is to be filtered off and washed and added to the main silica. The filtrate is reserved. The silica, thoroughly washed, is dried and ignited at a high temperature for twenty or thirty minutes. It is then weighed in a platinum crucible. After weighing it is treated with hydrofluoric acid and a little sulphuric, carefully evaporated and ignited strongly. The residue, which in extreme cases may amount to 2 or 3 per cent. of the rock, is weighed and deducted from the weight of the impure silica. It is retained in the crucible.
Alumina, &c.—The filtrate from silica is treated by the basic acetate method. That is, it is first treated by a cautious addition of a solution of soda, almost to the point of producing a precipitate, in order to neutralise the excess of acid; 2 or 3 grams of sodium acetate are added, and the whole boiled for a minute or so. The precipitate is filtered off and washed only slightly. Save the filtrate. The precipitate is dissolved in hydrochloric, or, perhaps better, in nitric acid; and is reprecipitated by adding an excess of ammonia and boiling. The precipitate is filtered and washed with water containing 2 per cent. of ammonium nitrate. Both filtrates are evaporated separately to a small bulk, a drop or two of ammonia being added to the second towards the finish. They are next filtered into a 6 or 8-ounce flask through a small filter, the second filtrate coming after, and serving in a manner as wash water for the first[113]. The two washed alumina precipitates are dried and placed in the platinum crucible containing the residue from silica after treatment with hydrofluoric acid. They are then ignited in an oxidising atmosphere at a high temperature for about 10 minutes. The weight, including that of the residue from the silica, is noted as that of "alumina, &c."
The weighed oxides are next fused with bisulphate of potash for some hours. The bisulphate should have been first fused, apart, until the effervescence from the escape of steam has stopped. The melt is dissolved out with cold water and dilute sulphuric acid, and any insoluble residue is filtered off, washed, ignited and weighed. The filtrate is reserved for determinations of iron and titanium. The residue, after weighing, may be treated[Pg 411] with hydrofluoric and sulphuric acids for any silica,[114] which would be determined by loss. It may be tested for barium sulphate by treatment with hot strong sulphuric acid; in which this salt dissolves, but is again insoluble (and so comes out as a white precipitate) on diluting with cold water; the acid also must be cold before adding the water. The filtrate containing the iron is reduced with sulphuretted hydrogen, boiled till free from that gas, filtered and titrated with a standard solution of permanganate of potassium. The iron found is calculated to ferric oxide by dividing by .7. The iron solution after titration serves for the determination of titanium oxide (TiO2). This is done colorimetrically, by adding peroxide of hydrogen free from hydrofluoric acid, and comparing the brown colour produced with that produced by the addition of a standard solution of titanium to an equal volume of water containing sulphuric acid.[115] The alumina is determined by difference. From the weight of the combined precipitate which has been recorded as "Alumina, &c.," deduct (1) the residue, insoluble, after fusion with bisulphate; (2) the ferric oxide; (3) the titanium oxide; and (4) the phosphoric oxide (P2O5), the amount of which is subsequently determined in a separate portion. This gives the alumina.
Manganous oxide, &c.—The filtrate from the "alumina, &c." contained in a 6 or 8-ounce flask, which it nearly fills, is made slightly alkaline with ammonia and treated with a small excess of ammonium sulphide; the flask is then corked and placed on one side for some time (a day or so) so that the manganese sulphide may separate. The precipitate is filtered off and washed with water containing ammonium chloride and a few drops of ammonium sulphide. The filtrate is reserved for lime, &c. The precipitate is digested with sulphuretted hydrogen water, to which one-fifth of its volume of strong hydrochloric acid has been added; this dissolves the sulphides of zinc and manganese; any black residue should be tested for copper and perhaps nickel. The solution is evaporated to dryness, taken up with a little water and treated with a small excess of solution of carbonate of soda. It is boiled and again evaporated, washed[Pg 412] out with hot water and filtered on to a small filter, dried, ignited, and weighed as Mn3O4. It is calculated to MnO. It may contain, and should be tested for oxide of zinc, which, if present, must be deducted. If the dish becomes stained during evaporation, take up with a few drops of hydrochloric and sulphurous acids, evaporate, and then treat with carbonate of soda.
Lime, &c.—The filtrate from the manganese sulphide is boiled, and without cooling, treated with ammonium oxalate in solution, which also should be heated to boiling. The liquid is filtered off and reserved for magnesia. The precipitate is dissolved in very little hydrochloric acid and reprecipitated by adding ammonium oxalate and ammonia to the boiling solution. The filtrate and washings from this are reserved for magnesia. The precipitate is either dissolved in dilute sulphuric and titrated with permanganate of potash as described under Lime (p. 322); or it is ignited and weighed as oxide. In this last case it may be examined for barium and strontium, the former of which will rarely be present.
Magnesia.—The filtrate from the first lime precipitate is treated with sodium phosphate and ammonia, and allowed to stand overnight. It is then filtered. The precipitate is dissolved in hydrochloric acid; the solution is filtered into the beaker containing the solution from the second lime precipitate. Ammonia and sodium phosphate are again added, and the precipitate, after standing, is filtered off, washed with water containing ammonia; it is then dried, ignited and weighed as magnesium pyrophosphate. This is calculated into magnesia.
Potash and Soda.—Weigh out .5 gram of the dried ore, and mix with an equal quantity of ammonic chloride; and to the mixture add gradually 4 grams of calcium carbonate ("precipitated"). Introduce into a platinum-crucible and cover loosely. Heat, at first, gently; and then at a red heat for from forty to sixty minutes. Transfer to a porcelain dish, and digest with 60 or 80 c.c. of water; warm and filter: to the filtrate add ammonic carbonate and ammonia, and filter; evaporate the filtrate to dryness, adding a few drops more of ammonic carbonate towards the end; when dry, heat gently, and then raise the temperature to a little below redness. Dissolve in a small quantity of water, add a drop of ammonic carbonate, and filter through a small filter into a weighed platinum dish. Evaporate, ignite gently, and weigh. The residue contains the soda and potash of the mineral as chlorides.
To determine the proportion of potassium, dissolve this residue in a little water, add platinum chloride in excess, evaporate to a[Pg 413] paste, extract with alcohol, decant through a small weighed filter, wash with alcohol, and dry at 100° C. Weigh. The substance is potassium platinic chloride (2KCl.PtCl4). Its weight, multiplied by 0.1941, will give the weight of potash (K2O).
To find the proportion of soda, multiply the weight of the potassium platinic chloride by 0.306; this gives the weight of potassium chloride. Deduct this from the weight of the mixed chlorides first got; the difference will be the sodium chloride, which weight, multiplied by 0.53, will give the weight of soda (Na2O).
Ferrous Oxide.—When a qualitative test shows both ferric and ferrous oxide to be present, the proportion of the ferrous oxide must be separately determined. The finely ground mineral mixed with dilute sulphuric acid is treated on a water bath with hydrofluoric acid. Solution is best effected in an atmosphere of carbonic acid. In about an hour the decomposition is complete, and the solution is diluted with cold water, and titrated with the solution of bichromate or of permanganate of potassium. The iron found is multiplied by 1.286, and reported as ferrous oxide. To find the proportion of ferric oxide, the ferrous iron found is multiplied by 1.428, and this is deducted from the weight of ferric oxide obtained by precipitation with ammonia. The ammonia precipitate contains the whole of the iron as ferric oxide; hence the necessity for calculating the ferrous oxide as ferric, and deducting it.
Phosphoric Oxide (P2O5).—Weigh up 5 grams of the finely-divided and dry sample, and digest with 10 or 20 c.c. of nitric acid; evaporate to dryness on the water-bath; take up with a little dilute nitric acid; dilute with water; and filter. Add a few grams of ammonic nitrate and 10 c.c. of ammonium molybdate solution, heat nearly to boiling, and allow to settle; filter off, and wash the yellow precipitate. Dissolve with dilute ammonia, add "magnesia mixture," and allow to stand overnight. Filter, wash with dilute ammonia, dry, ignite, and weigh as pyrophosphate of magnesia. The weight, multiplied by 0.6396, gives the weight of phosphoric oxide.
Soluble Silica.—Some silicates are acted on by hydrochloric acid, and leave on evaporation a residue; which, when the soluble salts have been washed out, consists generally of the separated silica with perhaps quartz and unattacked silicates. It should be ignited, weighed and boiled with a solution containing less than 10 per cent. of caustic soda: this dissolves the separated silica. The liquor is diluted, rendered faintly acid, and filtered. The residue is washed, ignited and weighed. The loss gives the soluble silica.[Pg 414]
Estimation of Silica in Slags (Ferrous silicates).—Take 1 gram of the powdered slag, treat with aqua regia, evaporate to dryness, extract with hydrochloric acid, filter, dry, ignite, and fuse the ignited residue with "fusion mixture," then separate and weigh the silica in the usual way. Slags are for the most part decomposed by boiling with aqua regia, but it will be found more convenient and accurate to first extract with acids and then to treat the residue as an insoluble silicate.
Estimation of "Silica and Insoluble Silicates" in an Ore.—Take 2 grams of the powdered mineral, evaporate with nitric acid (if sulphides are present), treat the dried residue (or the original substance if sulphides are absent) with 10 or 20 c.c. of hydrochloric acid; again evaporate to dryness, take up with dilute hydrochloric acid, filter, wash, ignite, and weigh.
Estimation of Silicon in Iron.—Place 2 grams of the metal (borings or filings) in a four-inch evaporating dish, and dissolve (with aid of heat) in 25 c.c. of dilute nitric acid. Evaporate to complete dryness, take up with 20 c.c. of hydrochloric acid, and allow to digest for one hour. Boil down to a small bulk, dilute with a 5 per cent. solution of hydrochloric acid, boil, and filter. Wash with acid and water, dry, ignite in a platinum crucible, and weigh the SiO2. This, multiplied by 0.4673, gives the weight of the silicon. The percentage is calculated in the usual way.
1. A certain rock is a mixture of 70 per cent. of quartz, 25 per cent. of potash-felspar, and 5 per cent. of potash-mica. What per cent. of silica will it contain?
2. Two grams of a mixture of silica and cassiterite left, after reduction in hydrogen, 1.78 grams. Assuming all the oxide of tin to have been reduced, what will be the percentage of silica?
3. The formula of a compound is 2FeO.SiO2. What percentage of silica will it contain?
4. Two grams of a sample of cast-iron gave 0.025 gram of silica. Find the percentage of silicon in the metal.
5. What weights of quartz and marble (CaCO3) would you take to make 30 grams of a slag having the formula CaO.SiO2?
Carbon compounds enter so largely into the structure of organised bodies that their chemistry is generally considered apart from that of the other elements under the head of Organic Chemistry. Carbon occurs, however, among minerals not only in the oxidised state (as carbonates), but also in the elementary form (as in diamond and graphite), and combined with hydrogen,[Pg 415] oxygen, &c. (as in petroleums, bitumens, lignites, shales, and coals). In small quantities "organic matter" is widely diffused in minerals and rocks. In shales and clays it may amount to as much as 10 or 20 per cent. (mainly as bituminous and coaly matters).
The assayer has only to take account of the organic matter when it is of commercial importance, so that in assays it is generally included under "loss on ignition."
In coals, shales, lignites, &c., the carbon compounds are, on heating, split up into oils and similar compounds. The products of distillation may be classified as water, gas, tars, coke, and ash. The assay of these bodies generally resolves itself into a distillation, and, in the case of the shales, an examination of the distillates for the useful oils, paraffin, creosote, &c., contained in them.
Elementary carbon is found in nature in three different forms, but these all re-act chemically in the same way. They combine with oxygen to form the dioxide.[116] The weight of oxygen required to burn a given weight of any form of carbon is the same, and the resulting product from all three has the same characteristic properties. Carbon dioxide is the common oxide of carbon. A lower oxide exists, but on burning it is converted into the dioxide. Wherever the oxidation of carbon takes place, if there is sufficient oxygen, carbon dioxide (carbonic acid) is formed; this re-action is the one used for the determination of carbon in bodies generally. The dioxide has acid properties, and combines with lime and other bases forming a series of salts called carbonates.
The carbon-compounds (other than carbonates, which will be subsequently considered) occurring in minerals are generally characterised by their sparing solubility in acids. The diamond is distinguished from other crystals by its hardness, lustre, and specific gravity. It may be subjected to a red heat without being apparently affected, but at a higher temperature it slowly burns away. Graphite, also, burns slowly, but at a lower temperature. The other bodies (coals, shales, &c.) differ considerably among themselves in the temperature at which they commence to burn. Some, such as anthracite, burn with little or no flame, but most give off gases, which burn with a luminous flame. They deflagrate when sprinkled on fused nitre, forming carbonate of potash. In making this test the student must remember that sulphur and, in fact, all oxidisable bodies similarly deflagrate, but it is only in the case of carbon compounds that carbonate of potash is formed. Carbon unites with iron and some of the metals to form carbides;[Pg 416] combined carbon of this kind is detected by the odour of the carburetted hydrogen evolved when the metal is treated with hydrochloric acid; for example, on dissolving steel in acid.
The natural carbon compounds, although, speaking generally, insoluble in hydrochloric or nitric acids, are more or less attacked by aqua regia. The assayer seldom requires these compounds to be in solution. The presence of "organic matter"[117] interferes with most of the reactions which are used for the determination of the metals. Consequently, in such cases, it should be removed by calcination unless it is known that its presence will not interfere. When calcination is not admissible it may be destroyed by heating with strong sulphuric acid and bichromate or permanganate of potash or by fusion with nitre.
Carbon may be separated from other substances by conversion into carbon dioxide by burning. In most cases substances soluble in acids are first removed, and the insoluble residue dried, weighed, and then calcined or burned in a current of air. The quantity of "organic matter" may be determined indirectly by the loss the substance undergoes, but it is better to determine the "organic carbon" by confining the calcination in a tube, and collecting and weighing the carbon dioxide formed. Each gram of carbon dioxide is equivalent to 0.2727 gram of carbon.

Instead of a current of oxygen or air, oxide of copper may be more conveniently used. The operation is as follows:—Take a clean and dry piece of combustion tube drawn out and closed at one end, as shown in the figure (fig. 70), and about eighteen inches long. Fit it with a perforated cork connected with a U-tube (containing freshly-fused calcium chloride in coarse grains) and a set of potash bulbs (fig. 71) (containing a strong solution of potash), the exit of which last is provided with a small tube containing calcium chloride or a stick of potash. Both the U-tube and bulbs should have a loop of fine wire, by which they may be suspended on the hook of the balance for convenience[Pg 417] in weighing. They must both be weighed before the combustion is commenced; to prevent absorption of moisture during weighing, &c., the ends are plugged with pieces of tube and glass rod.
Fill the combustion tube to a depth of about eight inches with some copper oxide, which has been recently ignited and cooled in a close vessel. Put in the weighed portion for assay and a little fresh copper oxide, and mix in the tube by means of an iron wire shaped at the end after the manner of a corkscrew. Put in some more oxide of copper, and clean the stirrer in it. Close loosely with a plug of recently ignited asbestos, place in the furnace, and connect the U-tube and bulbs in the way shown in the sketch (fig. 72).
See that the joints are tight, and then commence the combustion by lighting the burners nearest the U-tube; make the first three or four inches red hot, and gradually extend the heat backwards the length of the tube, but avoid too rapid a disengagement of gas. When gas ceases to come off, open the pointed end of the tube and draw a current of dried air through the apparatus.
The carbon dioxide is absorbed in the potash bulbs, and their increase in weight multiplied 0.2727 gives the amount of carbon in the substance taken.
The increase in weight in the calcium chloride tube will be due to the water formed by the oxidation of the combined hydrogen. If this last is required the increase in weight multiplied by 0.111 gives its amount.[Pg 418]
The determination of the actual carbon in coals and shales is seldom called for; if required, it would be performed in the way just described.[118] The ordinary assay of a sample of coal involves the following determinations—moisture, volatile matter, fixed carbon, ash, and sulphur. These are thus carried out:—
Determination of Moisture.—Take 3 grams of the powdered sample and dry in a water-bath for an hour or so. The loss is reported as moisture. Coals carry from 1 to 2 per cent. If the drying is carried too far, coals gain a little in weight owing to oxidation, so that it is not advisable to extend it over more than one or two hours.
Determination of Volatile Matter.—This determination is an approximate one, and it is only when working under the same conditions with regard to time, amount of coal taken, and degree of heat used, that concordant results can be arrived at. It is a matter of importance whether the coal has been previously dried before heating or not, since a difference of 2 per cent. may be got by working on the dried or undried sample. Take 2 grams of the powdered, but undried, sample of coal, place in a weighed platinum crucible, and support this over a good Bunsen burner by means of a thin platinum-wire triangle. The heat is continued until no further quantity of gas comes off and burns at the mouth. This takes only a few minutes. The cover is tightly fitted on, and when cold the crucible is weighed. The loss in weight, after deducting the moisture, gives the "volatile matter," and the residue consists of "fixed carbon" and "ash."
Determination of Ash.—The coke produced in the last operation is turned out into a porcelain dish and ignited over a Bunsen burner till the residue is free from particles of carbon. Calcination is hastened by stirring with a platinum wire. The operation may be done in a muffle, but this gives results a few tenths of a per cent. too low. The dish is cooled in a dessicator, and weighed. The increase in weight gives the amount of "ash," and the difference between this and the weight of the coke gives the "fixed carbon."
The assay is reported as follows:—
| Moisture at 100° C. | __ | per cent. |
| Volatile matter | __ | " |
| Fixed carbon | __ | " |
| Ash | __ | " |
| contains sulphur __ per cent. |
Determination of Sulphur.—The sulphur exists in the coal partly in organic combination, partly as metallic sulphide (iron pyrites, marcasite, &c.), and, perhaps, as sulphate. So that the sulphur determination must be separately reported, since a portion will go off with the volatile matter, and the remainder would be retained and weighed with the coke.
The sulphur is thus determined:—Take 1 gram of the coal and mix with 1.5 gram of a mixture of 2 parts of calcined magnesia and 1 part of carbonate of soda, and heat in a platinum crucible for one hour or until oxidation is complete. Turn out the mass and extract it with water and bromine, filter, acidulate with hydrochloric acid, boil off the bromine, and precipitate with baric chloride (estimating gravimetrically as given under Sulphur). Another method is as follows:—Take 1 gram of the coal and drop it gradually from a sheet of note paper on to 5 grams of fused nitre contained in a platinum dish. Extract with water, acidify with acetic acid, and estimate volumetrically as described under Sulphur.
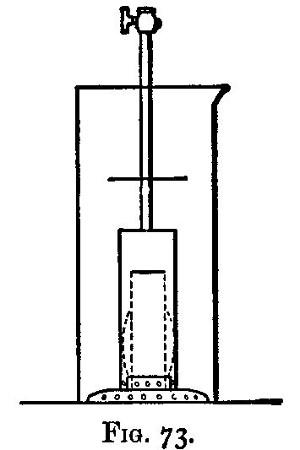
Calorific Effect of Coals.—The heat-giving value of a coal is best expressed in the number of pounds of water, previously heated to the boiling point, which it will convert into steam. This is generally termed its evaporative-power. It may be determined by means of the calorimeter (fig. 73). This consists of a glass cylinder marked to hold 29.010 grains of water. The instrument consists of a perforated copper stand, provided with a socket and three springs. The socket holds a copper cylinder which is charged with 30 grains of the dried coal mixed with 300 grains of a mixture of 3 parts of potassium chlorate and 1 part of nitre. The charge is well packed in the cylinder and provided with a small fuse of cotton saturated with nitre. Fill the glass cylinder to its mark with water and take the temperature with a thermometer marked in degrees Fahrenheit. Ignite the fuse and immediately cover with the outer copper cylinder (extinguisher-fashion), which will be held in its place by the springs. The stop-cock should be closed before this is done. Place the apparatus quickly in the cylinder of water. When the action is over open the stop-cock and agitate the water by raising and lowering the instrument a few times. Again take the temperature. The[Pg 420] rise in temperature, plus 10 per cent. for the heat used in warming the apparatus and lost by radiation, gives the evaporative-power.
The following is an example:—
| Temperature before experiment | 67.0° | F. |
| Temperature after " | 79.0° | " |
| ———— | ||
| Rise | 12.0° | " |
| + 1/10th | 1.2° | " |
| ———— | ||
| Gives | 13.2° | " |
One pound of the coal will evaporate 13.2 pounds of water.
The assay of these is carried out in the same way as that of coals, but the volatile matters are separately examined, and, in consequence, a larger quantity of material must be used. For the moisture, volatile matter, fixed carbon and ash, the determinations are the same, but a special distillation must be made to obtain a sufficient quantity of the volatile products for subsequent examination. Take 500 or 1000 grams of the well-sampled and powdered shale, and introduce into a cast-iron retort as shown in fig. 74. Lute the joint with fire-clay, place the cover on, and bolt it down. The bolts should have a covering of fire-clay to protect them from the action of the fire. Place the retort in a wind furnace, supporting it on a brick, and pack well around with coke. Build up the furnace around and over the retort with loose fire-bricks, and heat gradually.
As soon as water begins to drip, the tube of the retort is cooled by wrapping a wet cloth around it, and keeping wet with water. The water is kept from running into the receiver by a ring of damp fire-clay. A quantity of gas first comes over and will be lost, afterwards water and oily matters. The retort must be red hot at the close of the distillation, and when nothing more distils off, which occurs in about two or three hours, the wet cloth is removed, and the tube heated with a Bunsen burner to drive[Pg 421] forward the matter condensed in it into the receiver, and thus to clean the tube. It can be seen when the tube is clean by looking up through it into the red-hot retort. The receiver is then removed, and the retort, taken from the furnace, is allowed to cool. When cold it is opened, and the fixed carbon and ash weighed, as a check on the smaller assay.
The distillate of water and oil is warmed, and will separate into two layers, the upper one of which is oil, and the lower water. These are measured, and if the specific gravity of the oil is taken, its weight may be calculated. If the two liquids do not separate well, the water may be filtered off, after cooling, through a damped filter. The separation is, however, best effected in a separator (fig. 75). The liquids are poured into this, allowed to settle, and the lower layer drained off. The volume of the water is measured and its weight calculated in per cents. on the amount of shale taken.
Examination of the Oil.—A sufficient quantity of the oil must be got, so that if one distillation does not yield enough, the requisite quantity must be obtained by making two or more distillations. The oils are mixed, and the mixture, after having had its volume and specific gravity ascertained, is placed in a copper retort, and re-distilled with the aid of a current of steam. The residue in the retort is coke.
The distillate is separated from the water by means of the separator, and shaken for ten minutes with one-twentieth of its bulk of sulphuric acid (sp. g. 1.70). The temperature should not be allowed to rise above 40°. Allow to stand, and run off the "acid tar."
The oil is now shaken up with from 10 c.c. to 20 c.c. of sodic hydrate solution (sp. g. 1.3), allowed to stand, warmed for half-an-hour, and the "soda-tar" run off.
On mixing this soda-tar with dilute acid, the "crude shale oil creosote" separates, and is measured off.
The purified oil is next re-distilled in fractions, which come over in the following order:—"Naphtha," "light oil," "heavy oil," and "still bottoms." For the first product, which is only got from certain shales, the receiver is changed when the distillate has a specific gravity of 0.78. For the second product the process is continued till a drop of the distillate, caught as it falls from the neck of the retort on a cold spatula, shows signs of solidifying. This is "crude light oil."[Pg 422]
The receiver is changed, and the "heavy oil" comes over; towards the end a thick brown or yellow viscid product is got. The receiver is again changed, and the distillation carried to dryness.
The "crude light oil" is washed cold with 2 per cent. of sulphuric acid (concentrated), and afterwards with excess of soda. Thus purified it is again distilled to dryness, three fractions being collected as before. Naphtha, which is added to the main portion, and measured; "light oil," which is also measured; and "heavy oil," which is added to that got in the first distillation. This last is poured into a flat-bottom capsule, and allowed to cool slowly. The temperature may with advantage be carried below freezing-point. The cooled cake is pressed between folds of linen, and the paraffin scale detached and weighed.
The results may be reported thus:—
| Naphtha, sp. g. | —— |
| Light oil, sp. g. | —— |
| Heavy oil, sp. g. | —— |
| Paraffin scale | —— |
| Coke, &c. | —— |
The results are calculated in per cents. on the oil taken. Some workers take their fractions at each rise of 50° C. The composition of average shale, as given by Mills, is as follows:—Specific gravity, 1.877; moisture, 2.54.
| Gas | } | |
| Volatile matter, water, ammonia | } | 23.53 |
| Oil | } | |
| Fixed carbon | 12.69 | |
| Ash | 63.74 | |
| _____ | ||
| 99.96 |
The ash is made up of silica, 55.6; ferric oxide, 12.2; alumina, 22.14; lime, 1.5; sulphur, 0.9; soluble salts (containing 0.92 per cent. sulphuric oxide), 8.3.
| Total | sulphur | in shale | 1.8 | per cent. |
| " | " | in ash | 1.3 | " |
For further information on these assays, and for the assay of petroleums, bitumens, &c., the student is referred to Allen's "Commercial Organic Analysis," Vol. II.
Determination of Organic Carbon in a Limestone.—Take 1 or 2 grams and dissolve with a very slight excess of dilute hydrochloric acid, evaporate to dryness, and determine the carbon in the residue by combustion with copper oxide.[Pg 423]
Estimation of Carbon in a Sample of Graphite (Black-lead).—Weigh up 1 or 2 grams in a dish and calcine in the muffle till the carbon is burnt off. Weigh the residue, and calculate the carbon by difference.

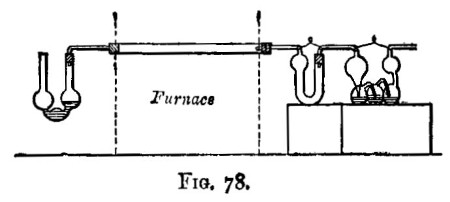
Determination of Carbon in Iron.—The carbon exists in two states—free (graphite) and combined. The following process estimates the total carbon:—The carbon existing as graphite may be separately estimated in another portion by the same process, but using hydrochloric acid to dissolve the iron instead of the copper solution:—Weigh up 2 grams of the iron (or a larger quantity if very poor in carbon), and attack it with 30 grams of ammonic-cupric chloride[119] dissolved in 100 c.c. of water. Let the reaction proceed for a quarter-of-an-hour, and then warm until the copper is dissolved. Allow to settle, and filter through a filtering-tube. This is a piece of combustion tube drawn out and narrowed at one end, as shown in fig. 76. The narrow part is blocked with a pea of baked clay, and on this is placed half-an-inch of silica sand (previously calcined to remove organic matter), then a small plug of asbestos, and then a quarter-of-an-inch of sand. The tube is connected with a pump working at a gentle pressure, and the solution is filtered through the tube with the aid of a small funnel (fig. 77). The residue is washed, first with dilute hydrochloric acid, and then with distilled water. The tube is dried by aspirating air through it, and gently warming with a Bunsen burner. The tube is then placed in a small combustion-furnace, and connected with calcium chloride and potash bulbs, as shown in fig. 78. The[Pg 424] potash bulb to the right of the figure must be weighed. A slow stream of air is drawn through the apparatus, and the heat gradually raised; in from thirty minutes to one hour the combustion will be complete. The potash bulbs are then disconnected and weighed, and the increase multiplied by 0.2727 gives the weight of carbon.
Carbon dioxide, which is formed by the complete oxidation of carbon, is a gas with a sweetish odour and taste, having a strong affinity for alkalies, and forming a series of compounds termed carbonates. The gas itself occurs in nature, and is sometimes met with in quantity in mining. The carbonates occur largely in nature, forming mountain masses of limestone, &c. Carbonates of many of the metals, such as carbonate of lead (cerussite), carbonate of iron (chalybite), carbonates of copper (malachite and chessylite), and carbonate of magnesia (magnesite), are common.
All the carbonates (those of the alkalies and alkaline earths excepted) are completely decomposed on ignition into the oxide of the metal and carbon dioxide; but the temperature required for this decomposition varies with the nature of the base. All carbonates are soluble with effervescence in dilute acids; some, such as chalybite and magnesite, require the aid of heat. The alkaline carbonates are soluble in water; the rest, with the exception of the bicarbonates, are insoluble therein.
Carbonates are recognised by their effervescence with acids—a stream of bubbles of gas are given off which collect in the tube, and possess the property of extinguishing a lighted match. The most characteristic test for the gas is a white precipitate, which is produced by passing it into lime or baryta-water, or into a solution of subacetate of lead.
The expulsion of carbon dioxide by the stronger acids serves for the separation of this body from the other acids and bases.
Dry Assay.—There is no dry assay in use. Any method which may be adopted will necessarily be applicable only to special compounds.
There are several methods in use which leave little to be desired either in speed or accuracy. We will give (1) a gravimetric method in which the estimation may be made directly by weighing the carbonic acid, or, indirectly, by estimating the carbon dioxide from the loss; (2) a volumetric one, by which an indirect determination[Pg 425] is made of the gas; and (3) a gasometric method, in which the volume of carbon dioxide given off is measured, and its weight deducted.
Direct Gravimetric Method.—Fit up the apparatus shown in the diagram (fig. 79). The various tubes are supported by a fixed rod with nails and wire loops, and connected by short lengths of rubber-tubing. The first tube contains soda-lime. The small flask is fitted with a rubber-stopper perforated with two holes, through one of which passes the tube of a pipette holding 25 or 30 c.c. This pipette is to contain the acid. The substance to be determined is weighed out into the flask. The second tube contains strong sulphuric acid; the third, pumice stone, saturated with copper sulphate solution, and dried until nearly white (at 200° C.); the fourth contains recently fused calcium chloride; and the fifth, which is the weighed tube in which the carbonic acid is absorbed, contains calcium chloride and soda-lime,[120] as shown in fig. 80. The sixth also contains calcium chloride and soda-lime; its object is to prevent the access of moisture and carbonic acid to the weighed tube from this direction; it is connected with an aspirator.
Having weighed the U-tube and got the apparatus in order, weigh up 1, 2, or 5 grams of the substance and place in the flask. Fill the pipette with dilute acid, close the clamp, and cork the flask. Then see that the apparatus is tight. Open the clamp[Pg 426] and allow from 10 to 20 c.c. of the acid to run on to the assay. Carbonic acid will be evolved and will be driven through the tubes. The gas should bubble through the sulphuric acid in a moderate and regular stream. When the effervescence slackens the clamp is opened and the greater part of the remaining acid run in. When the effervescence has ceased the clamp is opened to its full extent and a current of air drawn through with an aspirator. A gentle heat is applied to the flask; but it should not be prolonged or carried to boiling. After the removal of the heat a gentle current of air is drawn through the apparatus for 30 or 40 minutes. The weighed U-tube, which in the early part of the operation will have become warm if much carbonic acid was present, will by this time be cold. It is disconnected, plugged, and weighed. The increase in weight is due to the carbon dioxide of the sample.
Example.—Ore taken 1 gram.
| Weight | of tube, | before | 42.6525 | grams |
| " | " | after | 43.0940 | " |
| ———- | ||||
| Increase | equals CO2 | 0.4415 | " |
Indirect Gravimetric, or Determination by Loss.—Take a Geissler's carbonic-acid apparatus (fig. 81) and place in the double bulb some strong sulphuric acid. Put into the other bulb, the stopcock being closed, 3 or 4 c.c. of nitric acid diluted with water. Leave the apparatus in the balance-box for a few minutes and weigh. Introduce into the flask (through A) about 1 gram of the powdered substance and again weigh to find the exact amount added. Allow the acid to run gradually on to the carbonate, and when solution is complete, heat and aspirate. Cool and again weigh; the loss in weight is the carbonic acid.
For Example:—
| Weight | of apparatus | and acids | 85.494 | grams |
| " | " | marble | 86.879 | " |
| ——— | ||||
| Equal | to marble taken | 1.385 | " | |
| Weight | of apparatus | and marble | 86.879 | grams |
| " | " | minus carbonic acid | 86.2692 | " |
| ——— | ||||
| Equal | to carbonic acid | 0.6098 | " |
1.385 : 100 :: 0.6098 : x
x = 44.03 per cent.
The substance contains 44.03 per cent. of carbonic acid; a duplicate experiment gave 43.73 per cent.
This method is quicker, but less exact, than the direct gravimetric determination.
This, which is of somewhat limited application, is based upon the determination of the quantity of acid required to decompose the carbonate. It consists in adding to a weighed quantity of the mineral a known amount of standard solution of acid which is in excess of that required to effect the decomposition. The quantity of residual acid is then determined by titrating with standard solution of alkali. This method has been described under Lime.
This method is the quickest of all, and the least troublesome after the apparatus has been once prepared. It yields fairly accurate results when worked in the manner described below; but if greater precautions are taken the results are exact. It depends on the measurement of the volume of gas given off on treating the weighed sample with acid. The apparatus described, page 52, is used. Weigh out a portion of the mineral which shall contain not more than 0.15 gram of carbonic acid (or 0.4 gram of carbonate of lime) and put it in the bottle. Put in the inner tube 10 c.c. of dilute hydrochloric acid (1—1), cork tightly, and read off the level of the liquid in the burette after adjusting the pressure. Turn the acid over on to the mineral. Run out the water so as to keep the level in the two burettes the same. When effervescence has ceased, rotate the contents of the bottle; finally, adjust the level in the burettes and read off the volume. The increase in volume is due to the evolved carbon dioxide. At the same time read off the "volume corrector."
Some of the carbon dioxide remains dissolved in the acid in the generating bottle, and the quantity thus dissolved will depend on the amount of carbonate as well as on the amount of acid present. Consequently, a measured quantity of acid should be used in each assay and a comparative experiment made with a known weight of pure carbonate of lime which will yield about the same volume of gas. The number of c.c. of gas got in the assay multiplied by 4.7 will give the number of milligrams of pure carbonate of lime that must be taken for the standard. With ordinary work the error rarely exceeds half a c.c.[Pg 428]
The following example will illustrate the calculations:—
One gram of a mineral was taken, and yielded 49.0 c.c. of gas. The "volume corrector" reading was 100.4 c.c.
0.2405 gram of pure carbonate of lime was then taken, and treated in the same way; 50.5 c.c. of gas were got. The volume corrector still read 100.4 c.c.
0.2405 gram of carbonate of lime is equivalent to 0.1058 gram of carbon dioxide; then,
| 50.5 : 49.0 :: 0.1058 : x |
| x = 10.26 per cent. |
Estimation of Carbonic Acid in the Air of Mines.—According to a series of analyses by Angus Smith, the proportion of carbonic acid in the air of underground workings varied from 0.04 to 2.7 per cent. by volume. In places where men are working the proportion ought not to reach 0.25 per cent.
A simple method of determining whether a sample of air reaches this limit (0.25 per cent.) is described by Dr. C. Le Neve Foster in the "Proceedings of the Mining Association and Institute of Cornwall" for 1888. The apparatus used is an ordinary corked 8-ounce medicine bottle. This is filled with the air to be examined by sucking out its contents with a piece of rubber-tube. Half-an-ounce of dilute lime-water[121] (tinted with phenolphthalein) is poured in. If, on corking the bottle and shaking, the colour is not discharged, the air contains less than 0.25 per cent. of carbon dioxide. "If the colour fades slowly, and does not finally vanish till after a great deal of shaking, it may be assumed that the percentage of carbon dioxide does not greatly exceed one quarter; whereas, if the disappearance is rapid after a very few shakes, the contrary, of course, is the case." The dilute lime-water is measured out and carried in ordinary half-ounce phials. This method does not pretend to great accuracy, but as a method of distinguishing between good and bad air it is very convenient, and will be found useful.
For determining the actual proportion in the air the following plan is adopted:—Take a bottle which will hold about 50 ounces, and measure its capacity; fill the bottle with the air to be examined, pour in 100 c.c. of lime-water, and shake up for some time; add phenolphthalein, and titrate the remaining calcium hydrate with standard solution of oxalic acid.
The solution of oxalic acid is made by dissolving 2.25 grams of re-crystallised oxalic acid (H2C2O4.2H2O) in water and diluting to 1 litre. One c.c. = 0.001 gram of lime (CaO), or 0.0007857 gram of carbon dioxide.[Pg 429]
Take 100 c.c. of the same lime-water, to which add the same amount of phenolphthalein as before. Titrate. The difference between the two readings gives the amount of "acid" equivalent to the lime-water neutralised by the carbon dioxide. The number of c.c. thus used up, when multiplied by 0.3989, gives the number of c.c. of carbon dioxide (at 0° C. and 760 mm.) in the volume of air taken. This volume, which is that of the bottle less 100 c.c., must in accurate work be reduced to the normal temperature and pressure.[122] The percentage by volume can then be calculated.
1. In a gasometric determination 71.3 c.c. of gas were obtained from 0.2055 gram of mineral. The "volume corrector" reading was 102.2 c.c. 0.3445 gram of pure carbonate of lime gave 74.1 c.c. The "volume corrector" reading was 100.6. What is the percentage of carbon dioxide in the substance?
2. What volume of dry gas at 0° C. and 760 m.m. pressure should be obtained from 0.3445 gram of carbonate of lime? 1 c.c. of CO2 under these conditions weighs 1.97 milligrams.
3. A sample of coal is reported on as follows:—
| Specific gravity | 1.315 |
| Moisture | 1.001 |
| Volatile matter | 35.484 |
| Fixed carbon | 50.172 |
| Ash | 12.028 |
| ——— | |
| 100.000 |
What is there about this requiring explanation?
4. Calculate the percentage of carbonic acid in a mineral from the following data:—
| Weight | of apparatus | and acids | 87.0888 | grams | |
| " | " | " | plus mineral | 88.8858 | " |
| " | " | " | after loss of carbonic acid | 88.1000 | " |
5. A sample of pig iron contains 1.43 per cent. of "combined" and 2.02 per cent. of "free" carbon. Taking 2 grams of it for each determination, what weight of CO2 will be got on burning the residue from solution in ammonium cupric chloride, and what from the residue after solution in hydrochloric acid?
Boron occurs in nature as boric acid or sassoline (H3BO3); borax or tincal (Na2B4O7.10H2O); ulexite or boronatrocalcite (2CaB4O7.Na2B4O7); borocalcite (CaB4O7.4H2O); boracite, 2Mg3B8O15.MgCl2, and some other minerals. Boric acid is also a[Pg 430] constituent of certain silicates, such as tourmaline, axinite, and datholite.
The natural borates are used in the preparation of borax, which is largely employed as a preservative agent, for fluxing, and for other purposes.
There is only one series of boron compounds which have any importance. These are the borates in which the trioxide (B2O3) acts the part of a weak acid. The addition of any acid liberates boric acid, which separates out in cold solutions as a crystalline precipitate. Boric acid is soluble in alcohol and in hot water. On evaporating these solutions it is volatilised, although the anhydrous oxide is "fixed" at a red heat. The borates are mostly fusible compounds, and are soluble in acids and in solutions of ammonic salts.
Detection.—Boron in small quantities will escape detection unless specially looked for, but there is no difficulty in detecting its presence. Heated in the Bunsen-burner flame with "Turner's test," it gives an evanescent yellowish-green colour, due to fluoride of boron (BF3). "Turner's test" is a mixture of 5 parts of bisulphate of potash and 1 part of fluor spar. Boric acid itself imparts a characteristic green colour to the flame, which gives a spectrum made up of four well-marked and equidistant lines, three in the green and one in the blue. Solutions of boric acid give with "turmeric paper," which has been dipped into it and dried, a characteristic red tint. This is a very delicate test, but in trying it a blank experiment should be carried out alongside with a solution made up of the same re-agents which have been used in liberating the boric acid in the sample.
Solution and Separation.—The solution presents no difficulty, but the separation is troublesome. The best method is that of Gooch; who, if necessary, first fuses with carbonate of soda, and after the removal of chlorides and fluorides (by nitrate of silver or a lime salt), evaporates the aqueous extract with nitric or acetic acid to dryness in a retort and, subsequently, with repeated doses of 10 c.c. each of methyl alcohol. The distillate contains the boron as boric acid. Half a gram of the trioxide (B2O3) is completely carried over by two evaporations, each with 10 c.c. of the alcohol; but if water or foreign salts are present, more than this is required. In ordinary cases six such evaporations are sufficient for 0.2 gram of the oxide.[123]
Before the introduction of Gooch's process it was usual to determine the boron trioxide "by difference." If the alcoholic distillate containing the boric acid is digested with about 1 gram (a known weight) of lime for ten or fifteen minutes, the alcohol can be evaporated off without danger of loss. Either calcium nitrate or acetate (which will be formed at the same time) yields lime upon subsequent ignition. Consequently, the increase in weight, after ignition, upon that of the lime taken gives the amount of boron trioxide present. The trioxide contains 31.4 per cent. of boron (B). Since magnesia does not form a soluble hydrate it cannot satisfactorily be used instead of lime.
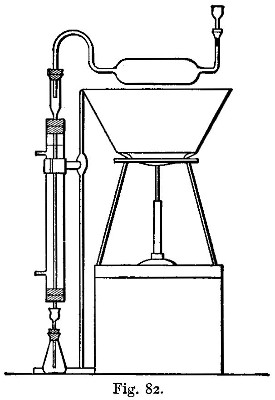
The apparatus required is shown in fig. 82. It consists of a small retort or evaporating vessel made out of a pipette of 200 c.c. capacity. This is heated by means of a paraffin-bath at 130° or 140° C. It is connected with an upright condenser, at the lower end of which is a small flask which serves as a receiver.
The quantity of the borate taken should contain not more than 0.2 gram of the trioxide. Insoluble compounds are "dissolved in nitric acid at once, or, if necessary, first fused with sodium carbonate." With soluble and alkaline borates sufficient nitric acid is added to render it faintly acid. The solution is then introduced into the retort.
"The lime, to retain the boric acid in the distillate, is ignited in the crucible in which the evaporation of the distillate is to be made subsequently." It is then cooled in the desiccator for ten minutes, and weighed. The lime is transferred to the receiving flask and slaked with a little water. The retort is lowered into the bath so that "only the rear dips below the surface." The[Pg 432] evaporation is carried to dryness, the retort being lowered further into the bath as the evaporation proceeds. Ten c.c. of methyl alcohol are introduced upon the residue, and the evaporation again started. Six such portions of alcohol are thus distilled and 2 c.c. of water are introduced and evaporated between the second and third, as also between the fourth and fifth distillations. If acetic acid is used instead of nitric in the first instance this addition of water is unnecessary.
The distillate is evaporated in the crucible ignited over the blowpipe, cooled in the desiccator for ten minutes and weighed. The increase in weight gives the boron trioxide. The results tend to be from 1 to 2 milligrams too high.
This method is applicable to the indirect determination of boric acid in borax and similar compounds. It is based on the measurement of the quantity of normal solution of acid required to replace the boric acid, and, consequently, is rather a measure of the soda present. The process is an alkalimetric one, and is carried out as follows:—Weigh up 3 grams of the sample and dissolve in water. Tint with methyl orange, and run in from an ordinary burette normal solution of sulphuric acid until a pink tint is got. 100 c.c. of the normal solution of acid are equal to 7.0 grams of boron trioxide (B2O3), or 10.1 grams of anhydrous borax (Na2B4O7).
Examination of Borax.—In addition to the determination just given, the following determinations are also required:—
Water.—Take about 2 grams and heat to tranquil fusion in a platinum crucible. Count the loss in weight as water.
Sulphuric Oxide.—Take 2 grams, dissolve in water, acidify with hydrochloric acid, filter, and precipitate with barium chloride. Wash the precipitate, ignite, and weigh as barium sulphate (see Sulphur).
Chlorine.—Take 2 grams, dissolve in water, acidify with nitric acid, filter, and add silver nitrate. Collect, wash, and weigh the precipitate as silver chloride.
Alumina.—Take 5 or 10 grams, dissolve in water, boil, add ammonia in slight excess, and filter off the precipitate when it has settled. Wash with hot water, ignite, and weigh as alumina (Al2O3).
[113] If the dishes show a manganese stain, wash them out with a few drops of hydrochloric and sulphurous acids. Pass the acid liquor through the same small filter but collect the liquor apart. Make ammoniacal and again pass through the filter, this time collecting the liquid with the main filtrate.
[114] This rarely amounts to more than 1 milligram.
[115] To make this, dissolve 1 gram of titanium oxide by fusing for some time with an excess of bisulphate of potash and dissolve out with cold water and sulphuric acid. Dilute to 1 litre, having previously added not less than 50 c.c. of strong sulphuric acid: 1 c.c. will contain .01 gram of TiO2. For the assay take 10 c.c. of this, add 2 c.c. of peroxide of hydrogen and dilute to 100 c.c. Run this from a burette into the flask until the colour equals that of the assay. Each c.c. equals 1 milligram of TiO2. Fluorides must be absent.
[116] C + O2 = CO2
[117] For example, soluble organic acids formed by partial oxidation with nitric acid.
[118] For coals, and other bodies containing sulphur, chromate of lead should be used instead of oxide of copper; and the temperature should be limited to dull redness.
[119] This may be prepared by dissolving 534 grams of ammonium chloride and 854 grams of crystallized cupric chloride (CuCl2.2H2O) in hot water and crystallizing.
[120] Soda-lime is made by dissolving 100 grams of "soda" in water, and carefully slaking 200 grams of lime with it. Evaporate to dryness in an iron dish and ignite at a low red heat in a crucible. Use the small lumps.
[121] Made by diluting 1 part by measure of saturated lime-water up to 10 with recently boiled distilled water.
[122] See under Gasometric Assays.
[123] See "A Method for the Separation and Estimation of Boric Acid," by F.A. Gooch, Chemical News, January 7, 1887.
| Symbols. | Names. | Atomic Weights. | Specific Gravity. | Melting Points. C. |
| Ag | Silver | 107.9 | 10.5 | 1000° |
| Al | Aluminium | 27.0 | 2.7 | 700° |
| As | Arsenic | 75.0 | 5.9 | |
| Au | Gold | 197.3 | 19.2 | 1200° |
| B | Boron | 11.0 | 2.7 | |
| Ba | Barium | 137.0 | 4.0 | |
| Be | Beryllium | 9.0 | 2.1 | |
| Bi | Bismuth | 208.9 | 9.8 | 270° |
| Br | Bromine | 80.0 | 3.2 | -25° |
| C | Carbon | 12.0 | ||
| Ca | Calcium | 40.0 | 1.6 | |
| Cd | Cadmium | 112.0 | 8.6 | 315° |
| Ce | Cerium | 140.2 | 6.7 | |
| Cl | Chlorine | 35.5 | ||
| Co | Cobalt | 59.0 | 8.5 | |
| Cr | Chromium | 52.1 | 7.3 | |
| Cs | Caesium | 132.9 | 1.9 | 25° |
| Cu | Copper | 63.4 | 8.9 | 1090° |
| Di | Didymium | 142.3 | 6.5 | |
| Er | Erbium | 166.3 | ||
| F | Fluorine | 19.0 | ||
| Fe | Iron | 56.0 | 7.8 | |
| Ga | Gallium | 69.0 | 5.9 | 30° |
| Ge | Germanium | 72.3 | ||
| H | Hydrogen | 1.0 | ||
| Hg | Mercury | 200.0 | 13.6 | -40° |
| I | Iodine | 126.8 | 4.9 | 106° |
| In | Indium | 113.7 | 7.4 | 175° |
| Ir | Iridium | 193.1 | 22.4 | |
| K | Potassium | 39.1 | 0.86 | 62.5° |
| La | Lanthanum | 138.2 | 6.1 |
| Symbols. | Names. | Atomic Weights. | Specific Gravity. | Melting Points. C. |
| Li | Lithium | 7.0 | 0.59 | 180° |
| Mg | Magnesium | 24.3 | 1.7 | |
| Mn | Manganese | 55.0 | 8.0 | |
| Mo | Molybdenum | 96.0 | 8.6 | |
| N | Nitrogen | 14.0 | ||
| Na | Sodium | 23.0 | 0.97 | 95.6° |
| Nb | Niobium | 94.0 | 4.1 | |
| Ni | Nickel | 58.7 | 8.9 | |
| O | Oxygen | 16.0 | ||
| Os | Osmium | 191.7 | 22.4 | |
| P | Phosphorus | 31.0 | 1.8 | 44° |
| Pb | Lead | 206.9 | 11.4 | 334° |
| Pd | Palladium | 106.6 | 11.4 | 1350° |
| Pt | Platinum | 195.0 | 21.5 | 2000° |
| Rb | Rubidium | 85.5 | 1.5 | 38.5° |
| Rh | Rhodium | 103.5 | 12.1 | |
| Ru | Ruthenium | 101.6 | 11.4 | |
| S | Sulphur | 32.0 | 2.0 | 115° |
| Sb | Antimony | 120.0 | 6.7 | 425° |
| Se | Selenium | 79.0 | 4.8 | 100° |
| Si | Silicon | 28.4 | 2.0 | |
| Sn | Tin | 119.0 | 7.3 | 235° |
| Sr | Strontium | 87.6 | 2.5 | |
| Ta | Tantalum | 182.6 | ||
| Te | Tellurium | 125.0 | 6.2 | 480° |
| Th | Thorium | 232.6 | 7.8 | |
| Ti | Titanium | 48.0 | 5.3 | |
| Tl | Thallium | 204.2 | 11.9 | 294° |
| U | Uranium | 239.6 | 18.4 | |
| V | Vanadium | 51.4 | 5.5 | |
| W | Tungsten | 184.0 | 19.1 | |
| Y | Yttrium | 89.1 | ||
| Yb | Ytterbium | 173.0 | ||
| Zn | Zinc | 65.3 | 6.9 | 423° |
| Zr | Zirconium | 90.6 | 4.1 |
The atomic weights in this table are in accord with the numbers given by F.W. Clarke (Dec. 6, 1890), chief chemist of the United States Geological Survey.[Pg 435]
 Table for Converting Degrees of the Centigrade
Thermometer into Degrees of Fahrenheit's Scale.
Table for Converting Degrees of the Centigrade
Thermometer into Degrees of Fahrenheit's Scale.
Nitric Acid.
Table showing the percentage, by Weight, of Real Acid (HNO3) in Aqueous Solutions of Nitric Acid of different Specific Gravities. Temperature, 15° C.
| 1.530 | 100.0 | 1.405 | 66.0 | 1.205 | 33.0 | ||
| 1.527 | 99.0 | 1.400 | 65.0 | 1.198 | 32.0 | ||
| 1.524 | 98.0 | 1.395 | 64.0 | 1.192 | 31.0 | ||
| 1.520 | 97.0 | 1.390 | 63.0 | 1.185 | 30.0 | ||
| 1.516 | 96.0 | 1.386 | 62.0 | 1.179 | 29.0 | ||
| 1.513 | 95.0 | 1.380 | 61.0 | 1.172 | 28.0 | ||
| 1.509 | 94.0 | 1.374 | 60.0 | 1.166 | 27.0 | ||
| 1.506 | 93.0 | 1.368 | 59.0 | 1.159 | 26.0 | ||
| 1.503 | 92.0 | 1.363 | 58.0 | 1.152 | 25.0 | ||
| 1.499 | 91.0 | 1.358 | 57.0 | 1.145 | 24.0 | ||
| 1.495 | 90.0 | 1.353 | 56.0 | 1.138 | 23.0 | ||
| 1.492 | 89.0 | 1.346 | 55.0 | 1.132 | 22.0 | ||
| 1.488 | 88.0 | 1.341 | 54.0 | 1.126 | 21.0 | ||
| 1.485 | 87.0 | 1.335 | 53.0 | 1.120 | 20.0 | ||
| 1.482 | 86.0 | 1.329 | 52.0 | 1.114 | 19.0 | ||
| 1.478 | 85.0 | 1.323 | 51.0 | 1.108 | 18.0 | ||
| 1.474 | 84.0 | 1.317 | 50.0 | 1.102 | 17.0 | ||
| 1.470 | 83.0 | 1.311 | 49.0 | 1.096 | 16.0 | ||
| 1.467 | 82.0 | 1.304 | 48.0 | 1.089 | 15.0 | ||
| 1.463 | 81.0 | 1.298 | 47.0 | 1.083 | 14.0 | ||
| 1.460 | 80.0 | 1.291 | 46.0 | 1.077 | 13.0 | ||
| 1.456 | 79.0 | 1.284 | 45.0 | 1.071 | 12.0 | ||
| 1.452 | 78.0 | 1.277 | 44.0 | 1.065 | 11.0 | ||
| 1.449 | 77.0 | 1.270 | 43.0 | 1.060 | 10.0 | ||
| 1.445 | 76.0 | 1.264 | 42.0 | 1.053 | 9.0 | ||
| 1.442 | 75.0 | 1.257 | 41.0 | 1.047 | 8.0 | ||
| 1.438 | 74.0 | 1.251 | 40.0 | 1.041 | 7.0 | ||
| 1.435 | 73.0 | 1.244 | 39.0 | 1.034 | 6.0 | ||
| 1.431 | 72.0 | 1.238 | 38.0 | 1.028 | 5.0 | ||
| 1.427 | 71.0 | 1.232 | 37.0 | 1.022 | 4.0 | ||
| 1.423 | 70.0 | 1.225 | 36.0 | 1.016 | 3.0 | ||
| 1.418 | 69.0 | 1.218 | 35.0 | 1.010 | 2.0 | ||
| 1.414 | 68.0 | 1.212 | 34.0 | 1.004 | 1.0 | ||
| 1.410 | 67.0 |
Table showing the percentage, by Weight, of Real Acid (HCl) in Aqueous Solutions of Hydrochloric Acid of different Specific Gravities. Temperature, 15° C.
| 1.2000 | 40.78 | 1.1410 | 28.54 | 1.0798 | 16.31 | ||
| 1.1982 | 40.37 | 1.1389 | 28.13 | 1.0778 | 15.90 | ||
| 1.1964 | 39.96 | 1.1369 | 27.72 | 1.0758 | 15.49 | ||
| 1.1946 | 39.55 | 1.1349 | 27.32 | 1.0738 | 15.08 | ||
| 1.1928 | 39.14 | 1.1328 | 26.91 | 1.0718 | 14.68 | ||
| 1.1910 | 38.74 | 1.1308 | 26.50 | 1.0697 | 14.27 | ||
| 1.1893 | 38.33 | 1.1287 | 26.10 | 1.0677 | 13.86 | ||
| 1.1875 | 37.92 | 1.1267 | 25.69 | 1.0657 | 13.45 | ||
| 1.1857 | 37.51 | 1.1247 | 25.28 | 1.0637 | 13.05 | ||
| 1.1846 | 37.11 | 1.1226 | 24.87 | 1.0617 | 12.64 | ||
| 1.1822 | 36.70 | 1.1206 | 24.46 | 1.0597 | 12.23 | ||
| 1.1802 | 36.29 | 1.1185 | 24.06 | 1.0577 | 11.82 | ||
| 1.1782 | 35.88 | 1.1164 | 23.65 | 1.0557 | 11.41 | ||
| 1.1762 | 35.47 | 1.1143 | 23.24 | 1.0537 | 11.01 | ||
| 1.1741 | 35.07 | 1.1123 | 22.83 | 1.0517 | 10.60 | ||
| 1.1721 | 34.66 | 1.1102 | 22.43 | 1.0497 | 10.19 | ||
| 1.1701 | 34.25 | 1.1082 | 22.02 | 1.0477 | 9.78 | ||
| 1.1681 | 33.84 | 1.1061 | 21.61 | 1.0457 | 9.38 | ||
| 1.1661 | 33.43 | 1.1041 | 21.20 | 1.0437 | 8.97 | ||
| 1.1641 | 33.03 | 1.1020 | 20.79 | 1.0417 | 8.56 | ||
| 1.1620 | 32.62 | 1.1000 | 20.39 | 1.0397 | 8.15 | ||
| 1.1599 | 32.21 | 1.0980 | 19.98 | 1.0377 | 7.75 | ||
| 1.1578 | 31.80 | 1.0960 | 19.57 | 1.0357 | 7.34 | ||
| 1.1557 | 31.40 | 1.0939 | 19.16 | 1.0337 | 6.93 | ||
| 1.1536 | 30.99 | 1.0919 | 18.76 | 1.0318 | 6.52 | ||
| 1.1515 | 30.58 | 1.0899 | 18.35 | 1.0298 | 6.11 | ||
| 1.1494 | 30.17 | 1.0879 | 17.94 | 1.0279 | 5.51 | ||
| 1.1473 | 29.76 | 1.0859 | 17.53 | 1.0259 | 5.30 | ||
| 1.1452 | 29.36 | 1.0838 | 17.12 | 1.0239 | 4.89 | ||
| 1.1431 | 28.95 | 1.0818 | 16.72 | 1.0200 | 4.01 |
Table showing the percentage, by Weight, of Real Ammonia (NH3) in Aqueous Solutions of Ammonia of different Specific Gravities. Temperature, 14° C.
| 0.8844 | 36.0 | 0.9145 | 23.6 | 0.9534 | 11.6 | ||
| 0.8852 | 35.6 | 0.9156 | 23.2 | 0.9549 | 11.2 | ||
| 0.8860 | 35.2 | 0.9168 | 22.8 | 0.9563 | 10.8 | ||
| 0.8868 | 34.8 | 0.9180 | 22.4 | 0.9578 | 10.4 | ||
| 0.8877 | 34.4 | 0.9191 | 22.0 | 0.9593 | 10.0 | ||
| 0.8885 | 34.0 | 0.9203 | 21.6 | 0.9608 | 9.6 | ||
| 0.8894 | 33.6 | 0.9215 | 21.2 | 0.9623 | 9.2 | ||
| 0.8903 | 33.2 | 0.9227 | 20.8 | 0.9639 | 8.8 | ||
| 0.8911 | 32.8 | 0.9239 | 20.4 | 0.9654 | 8.4 | ||
| 0.8920 | 32.4 | 0.9251 | 20.0 | 0.9670 | 8.0 | ||
| 0.8929 | 32.0 | 0.9264 | 19.6 | 0.9685 | 7.6 | ||
| 0.8938 | 31.6 | 0.9277 | 19.2 | 0.9701 | 7.2 | ||
| 0.8948 | 31.2 | 0.9289 | 18.8 | 0.9717 | 6.8 | ||
| 0.8957 | 30.8 | 0.9302 | 18.4 | 0.9733 | 6.4 | ||
| 0.8967 | 30.4 | 0.9314 | 18.0 | 0.9749 | 6.0 | ||
| 0.8976 | 30.0 | 0.9327 | 17.6 | 0.9765 | 5.6 | ||
| 0.8986 | 29.6 | 0.9340 | 17.2 | 0.9781 | 5.2 | ||
| 0.8996 | 29.2 | 0.9353 | 16.8 | 0.9790 | 4.8 | ||
| 0.9006 | 28.8 | 0.9366 | 16.4 | 0.9807 | 4.6 | ||
| 0.9016 | 28.4 | 0.9380 | 16.0 | 0.9823 | 4.2 | ||
| 0.9026 | 28.0 | 0.9393 | 15.6 | 0.9839 | 3.8 | ||
| 0.9036 | 27.6 | 0.9407 | 15.2 | 0.9855 | 3.4 | ||
| 0.9047 | 27.2 | 0.9420 | 14.8 | 0.9873 | 3.0 | ||
| 0.9057 | 26.8 | 0.9434 | 14.4 | 0.9890 | 2.6 | ||
| 0.9068 | 26.4 | 0.9449 | 14.0 | 0.9907 | 2.2 | ||
| 0.9078 | 26.0 | 0.9463 | 13.6 | 0.9924 | 1.8 | ||
| 0.9089 | 25.6 | 0.9477 | 13.2 | 0.9941 | 1.4 | ||
| 0.9100 | 25.2 | 0.9491 | 12.8 | 0.9959 | 1.0 | ||
| 0.9111 | 24.8 | 0.9505 | 12.4 | 0.9975 | 0.6 | ||
| 0.9122 | 24.4 | 0.9520 | 12.0 | 0.9991 | 0.2 | ||
| 0.9133 | 24.0 |
Table showing the percentage, by Weight, of Real Acid (H2SO4) in Aqueous Solutions of Sulphuric Acid of varying Specific Gravity. Temperature, 15° C.
| 1.838 | 100.0 | 1.568 | 66.0 | 1.247 | 33.0 | ||
| 1.840 | 99.0 | 1.557 | 65.0 | 1.239 | 32.0 | ||
| 1.841 | 98.0 | 1.545 | 64.0 | 1.231 | 31.0 | ||
| 1.841 | 97.0 | 1.534 | 63.0 | 1.223 | 30.0 | ||
| 1.840 | 96.0 | 1.523 | 62.0 | 1.215 | 29.0 | ||
| 1.838 | 95.0 | 1.512 | 61.0 | 1.206 | 28.0 | ||
| 1.836 | 94.0 | 1.501 | 60.0 | 1.198 | 27.0 | ||
| 1.834 | 93.0 | 1.490 | 59.0 | 1.190 | 26.0 | ||
| 1.831 | 92.0 | 1.480 | 58.0 | 1.182 | 25.0 | ||
| 1.827 | 91.0 | 1.469 | 57.0 | 1.174 | 24.0 | ||
| 1.822 | 90.0 | 1.458 | 56.0 | 1.167 | 23.0 | ||
| 1.816 | 89.0 | 1.448 | 55.0 | 1.159 | 22.0 | ||
| 1.809 | 88.0 | 1.438 | 54.0 | 1.151 | 21.0 | ||
| 1.802 | 87.0 | 1.428 | 53.0 | 1.144 | 20.0 | ||
| 1.794 | 86.0 | 1.418 | 52.0 | 1.136 | 19.0 | ||
| 1.786 | 85.0 | 1.408 | 51.0 | 1.129 | 18.0 | ||
| 1.777 | 84.0 | 1.398 | 50.0 | 1.121 | 17.0 | ||
| 1.767 | 83.0 | 1.388 | 49.0 | 1.113 | 16.0 | ||
| 1.756 | 82.0 | 1.379 | 48.0 | 1.106 | 15.0 | ||
| 1.745 | 81.0 | 1.370 | 47.0 | 1.098 | 14.0 | ||
| 1.734 | 80.0 | 1.361 | 46.0 | 1.091 | 13.0 | ||
| 1.722 | 79.0 | 1.351 | 45.0 | 1.083 | 12.0 | ||
| 1.710 | 78.0 | 1.342 | 44.0 | 1.075 | 11.0 | ||
| 1.698 | 77.0 | 1.333 | 43.0 | 1.068 | 10.0 | ||
| 1.686 | 76.0 | 1.324 | 42.0 | 1.061 | 9.0 | ||
| 1.675 | 75.0 | 1.315 | 41.0 | 1.053 | 8.0 | ||
| 1.663 | 74.0 | 1.306 | 40.0 | 1.046 | 7.0 | ||
| 1.651 | 73.0 | 1.297 | 39.0 | 1.039 | 6.0 | ||
| 1.639 | 72.0 | 1.289 | 38.0 | 1.032 | 5.0 | ||
| 1.627 | 71.0 | 1.281 | 37.0 | 1.025 | 4.0 | ||
| 1.615 | 70.0 | 1.272 | 36.0 | 1.019 | 3.0 | ||
| 1.604 | 69.0 | 1.264 | 35.0 | 1.013 | 2.0 | ||
| 1.592 | 68.0 | 1.256 | 34.0 | 1.006 | 1.0 | ||
| 1.580 | 67.0 |
In the case of small buttons of gold the weight can be determined more easily and accurately by measuring with the help of a microscope than by the actual use of a balance. Moreover, the method of measurement is applicable to the determination of quantities of gold too minute to affect even the most delicate balance.
For quantities of gold of from .5 to .005 milligram a microscope with 1/2 inch objective and B eyepiece is suitable. The measurements are made with the help of a scale engraved (or, better, photographed) on a circular piece of glass which rests on the diaphragm of the eyepiece. This scale and the object upon the stage can be easily brought into focus at the same time. The button of gold obtained by cupelling is loosened from the cupel by gently touching with the moistened point of a knife; it generally adheres to the knife, and is then transferred to a glass slide. The slide is placed on the stage of the microscope, illuminated from below; and the button is brought into focus, and so placed that it apparently coincides with the scale. The diameters in two or three directions (avoiding the flattened surface) are then read off: the different directions being got by rotating the eyepiece. The mean diameter is taken. The weight of the button is arrived at by comparing with the mean diameter of a standard prill of gold of known weight. The weights are in the proportion of the cubes of the diameters. For example, suppose a prill has been obtained which measures 12.5 divisions of the scale, and that a standard prill weighing 0.1 milligram measures 11.1 divisions. The weight will be calculated as follows:
11.13 : 12.53 :: 0.1 : x
| 0.1×12.5×12.5×12.5 | ||
| x = | —————————— | = 0.143 milligram. |
| 11.1×11.1×11.1 |
The calculations are simplified by the use of a table of cubes. The standard prills used in the comparison should not differ much in size from[Pg 441] the prills to be determined. They are prepared by alloying known weights of gold and lead, so as to get an alloy of known composition, say one per cent. gold. Portions of the alloy containing the weight of gold required (say 0.1 milligram) are then weighed off and cupelled on small smooth cupels, made with the finest bone-ash. Care must be taken to remove the cupels as soon as cupellation has finished. Several standard prills of the same size should be made at the same time, and their mean diameter calculated. The lead for making the gold-lead alloy is prepared from litharge purified by reducing from it about 10 per cent. of its lead by fusion with a suitable proportion of flour; the purified litharge is powdered, mixed with sufficient flour and reduced to metal.
In determining the gold contained in small buttons of silver-gold alloy obtained in assaying (and in which the silver is almost sure to be in excess of that required for parting), transfer the button from the cupel to a small clean porcelain crucible; pour on it a drop or two of nitric acid (diluted with half its bulk of water), and heat gently and cautiously until action has ceased. If the residual gold is broken up, move the crucible so as to bring the particles together, so that they may cohere. Wash three or four times with distilled water, about half filling the crucible each time and decanting off against the finger. Dry the crucible in a warm place; and when dry, but whilst still black, take the gold up on a small piece of pure lead. Half a grain of lead is sufficient, and it is best to hold it on the point of a blunt penknife, and press it on the gold in the crucible. The latter generally adheres. Transfer to a small smooth cupel and place in the muffle. When the cupellation has finished, the button of gold is measured as already described.
For the following remarks and experiments we are indebted to Mr. J.W. Westmoreland, who has had considerable experience with the process. Having dissolved the ore he converts the metals into sulphates by evaporating with sulphuric acid. The copper is then separated as subsulphide by means of hyposulphite of soda, and the precipitate is washed, dried, and calcined. The resulting oxide of copper is then dissolved in nitric acid; and to the concentrated solution, a saturated solution of carbonate of soda is added in sufficient quantity to throw down a considerable proportion of the copper. Acetic acid is added to dissolve the precipitate, and when this is effected more of the acid is poured on so as to render the solution strongly acid. To this potassium iodide crystals are added in the proportion of ten parts of iodide to each one part of copper supposed to be present. The solution is then titrated with "hypo" as usual.
For the examination of technical products experiments made in sulphuric acid solutions have no value, since arsenic acid, which is generally present to a greater or less extent, affects the end reaction. In such solutions bismuth may also interfere.
The solution best suited for the assay is one containing acetate of soda and free acetic acid. The presence of acetate of soda counteracts the interference of arsenic and of bismuth.
The return of the blue colour after titration is due to the excessive[Pg 442] dilution of the assay, or to an insufficiency of potassium iodide, or to the presence of nitrous fumes. The interference of an excess of sodium acetate is avoided by adding more iodide crystals to the extent of doubling the usual amount.
The interference of lead can be avoided by the addition of sulphuric acid or of phosphate of soda to the acid solution containing the copper, and before neutralising with carbonate of soda. The end reaction is, however, with care distinguishable without this addition. The following experiments, each containing .0648 gram of lead, were made by him in illustration:
| Copper taken. | Reagent added. | Copper found. | End reaction. |
| .2092 gram | — | .2077 gram | fairly satisfactory |
| .2101 " | — | .2092 " | " |
| .2167 " | sulphuric acid | .2152 " | " |
| .2117 " | " | .2108 " | " |
| .2109 " | phosphate of soda | .2092 " | good, colourless |
| .2205 " | " | .2174 " | rather yellow |
Effect of Sodium Acetate.—Each solution contained .3343 gram of copper.
| a.b.c. | d. | e. | f. | g. | |
| grams. | grams. | grams. | grams. | grams. | |
| "Acetate" added | — | 16.2 | 16.2 | 16.2 | 16.2 |
| "Iodide" added | 3.5 | 3.5 | 7.0 | 3.5 | 7.0 |
| Copper found | .3343 | .3324 | .3351 | .3269 | .3356 |
In these experiments, except with the excessive quantities of acetate of soda and the insufficiency of potassium iodide in the cases of d and f, there was no difficulty with the after-blueing.
The following method of separating and estimating cobalt and nickel has been described by Mr. James Hope,[125] with whom it has been in daily use for several years with completely satisfactory results.
The quantity of ore taken should contain about .5 gram of the mixed metals. It is dissolved in hydrochloric acid or aqua regia, and the solution evaporated to dryness. The residue is taken up with dilute hydrochloric acid and hot water. The solution is filtered off from the silica, freed from second group metals by treatment with sulphuretted hydrogen and filtered, and after oxidation with nitric acid is separated from iron and alumina by the basic acetate method (page 233). The precipitate is redissolved in a little hydrochloric acid, and again precipitated by sodium acetate. The two filtrates are mixed and treated with a little acetic acid, and the cobalt and nickel are then precipitated as sulphides by a current of sulphuretted hydrogen. The precipitate is filtered off, washed, dried,[Pg 443] and calcined, and the resulting oxides are weighed to get an idea as to the quantity of the two metals present.
The calcined precipitate is dissolved in a small covered beaker in aqua regia with the help of a few drops of bromine to remove any separated sulphur, and the solution evaporated to dryness with a few drops of sulphuric acid. The residue is dissolved in hot water, diluted to about 50 c.c., and heated to boiling. About 2 grams (four times the quantity of mixed metals present) of ammonium phosphate (AmH2PO4) are weighed off, dissolved in the smallest possible quantity of water, and boiled for a minute or two with a few c.c. of dilute sulphuric acid. This is added to the boiling-hot solution of cobalt and nickel, which is then treated cautiously with dilute ammonia until the precipitate partially dissolves. The addition of the ammonia is continued drop by drop with constant stirring, until the cobalt comes down as a pink precipitate of ammonium cobalt phosphate (AmCoPO4). The beaker is placed on the top of a water bath with occasional stirring for five or ten minutes. The blue liquid containing the nickel is decanted through a small filter and the precipitate is dissolved with a few drops of dilute sulphuric acid. The resulting solution is treated with a small excess of ammonium phosphate and the cobalt again precipitated by the cautious addition of ammonia exactly as before. The precipitate containing the whole of the cobalt is filtered off and washed with small quantities of hot water. The filtrate is added to the previous one containing the greater part of the nickel.
The ammonium cobalt phosphate is dried, transferred to a platinum crucible, and ignited over a Bunsen flame for fifteen or twenty minutes. A purple coloured cobalt pyrophosphate (Co2P2O7) is thus formed, and is weighed. It contains 40.3 per cent. of cobalt.
The mixed filtrates containing the nickel are placed in a tall beaker, and dilated if necessary to about 200 c.c. Ten c.c. of strong ammonia are added, and the solution, heated to 70° C., is ready for electrolysis. A battery of two 1-1/2 pint Bunsen cells is used. This is found capable of depositing from .15 to .20 gram of nickel per hour, and from two to three hours is generally sufficient for the electrolysis. The electrode with the deposited nickel is washed with distilled water, afterwards with alcohol as described under copper, and is then dried and weighed.
The following results obtained with this method by Mr. Hope illustrate the accuracy of the method. They were obtained by working on solutions containing known weights of the two metals:
| Taken. | Found. | ||
| Cobalt. | Nickel. | Cobalt. | Nickel. |
| .1236 gram | .1155 gram | .1242 gram | .1155 gram |
| .1236 " | .0577 " | .1232 " | .0575 " |
| .2472 " | .0577 " | .2449 " | .0585 " |
| .3708 " | .0577 " | .3701 " | .0580 " |
| .0618 " | .3465 " | .0619 " | .3454 " |
| .0618 " | .2310 " | .0625 " | .2295 " |
| .0618 " | .1155 " | .0621 " | .1155 " |
[124] For fuller information see a paper on "The Estimation of Minute Quantities of Gold," by Dr. George Tate; read before the Liverpool Polytechnic Society, Nov. 1889.
[125] Journal of the Society of Chemical Industry, No 4, vol. ix. April 30, 1890.
The problem of the sampler is essentially the same as that of the student of statistics. One aims at getting a small parcel of ore, the other a number of data, but each hopes to obtain what shall represent a true average applicable to a much larger mass of material. Ignoring the mechanical part of the problems, the sampling errors of the one and the deviations from the average of the other are the same thing.
It may be doubted whether many not specially trained in the study of statistics could answer such a question as the following:—Seven hundred thousand men being employed, there are, in a given year, one thousand deaths from accident. Assuming the conditions to remain unaltered, within what limits could one foretell the number of deaths by accident in any other year?
On the other hand, there is a widespread belief in the efficacy of what is called the law of averages. Even the ordinary newspaper reader is accustomed to look on the national death-rate or birth-rate as a thing capable of being stated with accuracy to one or two places of decimals, and he knows that the annual number of suicides is practically constant.
If a man played whist often and kept a record of the number of trumps n each hand, he would find fortune treated him quite fairly; in a year's play the average number would deviate very little from the theoretical average, i.e., one-quarter of thirteen. And a knowledge of this truth is useful, and that not merely in keeping ejaculations in due restraint. But every good player knows more than this: he has a sense of what variations in the number of trumps may reasonably be expected. For example, he will be prepared to risk something on neither of his opponents having more than five trumps, and will accept it as a practical certainty that no one has more than eight. Much of what is known as good judgment is based on a proper estimate of deviations from the average. The question has an important bearing on sampling, as may be seen from the fact that shuffling and dealing at cards are but modifications of the well-known mixing and quartering of the sampler.
Because of this bearing on sampling and for other reasons, I became many years ago much interested in the question, and gave to its solution perhaps more labour than it was worth. In books on Medical Statistics[Pg 445] the answer to the question is stated in a mathematical formula, called Poisson's formula, which, in a modified form, I shall give further on. But this did not satisfy me, because I wanted to learn what a reasonably safe limit of error actually meant, and this could be best learnt by experiment; so with the help of some friends I went in for a thorough course of penny-tossing.
Tossing a penny twenty times, an average result would be ten heads and ten tails. To find the deviations from this, we tossed two hundred twenties, i.e., four thousand times. Of the two hundred, thirty-three gave the exact average, viz.:—10 heads; sixty-four gave an error of one, viz.:—9 or 11 heads; forty-nine, an error of two; twenty-six, an error of three; twenty, an error of four; eight gave an error of five, and this limit was not exceeded. From these we may say that six is a reasonably safe limit of error. Ninety-seven cases, say one-half, gave an error not exceeding one; and the mean error is 1.8.
In other words, in twenty tosses you will not get more than 16 nor less than 4 heads; you are as likely as not to get 9, 10, or 11 heads; and lastly, if you lost in twenty throws all heads or tails over 10 your average loss would be 1.8 penny, or say roughly 2d. on the twenty throws.
It was necessary to compare these with another series containing a larger average, say that of 100 heads in 200 throws. I confess the labour of tossing pennies two hundred at a time was little to our taste. So from a bag of pennies borrowed from the bank, we weighed out samples containing two hundred, and for an evening we were busy counting heads and tails in these. The heads in sixty samples ranged from 80 to 114. One hundred heads occurred seven times. The extent and frequency of the errors is shown in the table.
| Error. | No. of Times. | Error. | No. of Times. | Error. | No. of Times. |
| 1 | 8 | 6 | 3 | 11 | 1 |
| 2 | 5 | 7 | 3 | 14 | 3 |
| 3 | 6 | 8 | 3 | 15 | 1 |
| 4 | 3 | 9 | 7 | 18 | 2 |
| 5 | 6 | 10 | 1 | 20 | 1 |
We may call the limit of error 21. Twenty-nine results out of sixty, say one-half, had an error not exceeding 4; and the mean error is 5.6. In comparing these with the series 10 in 20 we must, working by rule, divide not by 10 but by 3.16, the square root of 10; for if we multiply an average by any number[126] the error is also multiplied but only by the square root of the number. The error varies as the square root of the number. Now
| 21/3.16 = 6.6 = limit of error for 10 in 20. |
| 5.6/3.16 = 1.8 = mean error " " " |
| 4/3.16 = 1.2 = probable error " " " |
It will be seen that these calculated results agree fairly well with those actually obtained. The rule by which these calculations are made is important[Pg 446] and will bear further illustration. To calculate the number of heads in 3200 throws, we have to find the limit of error on a true average of 1600 in 3200. This being 16 times the average of 100 in 200, the corresponding errors must be multiplied by 4. This gives
21×4 = 84 = limit of error.
5.6×4 = 22.4 = mean error.
4×4 = 16 = probable error.
The results I have actually obtained with these large numbers are hardly enough to base much on, but have a value by way of confirmation. Expecting 1600 heads, the actual numbers were 1560, 1596, 1643, 1557, 1591, 1605, 1615, 1545.
It will be seen that exactly half are within the probable error; but this, considering the small number of results, must be more or less of an accident; it is more to the point they are all well within the limits of error.
I have a large number of other results which with a single exception are all in accord with those given; and this exception only just overstepped the limits. It was like a case of nine trumps, which though in a sense possible, is very unlikely to happen in any one's experience.
But even now we are not quite in a position to answer the question with which we started. If you refer to it you will see that we are face to face with this problem: the limit of variation on the 1000 who died would be say 70,[127] ignoring decimals. But if we calculate on the number who did not die, viz.—699,000,[128] we shall get a variation 26 times as great as this. But it is evident the variation must be the same in each case. I submitted this kind of problem also to the test of experiment, the results of which gave me great faith in Poisson's formula.
Imagine two hundred pennies in a bag all heads up. Any shaking will spoil this arrangement and give a certain proportion of tails. And, further, the probable effect of shaking and turning will be to reduce the preponderance of heads or tails whichever may be in excess. This of course is the reason why we are so unlikely to get more than 120 of them in either position.
But if the two hundred pennies are increased to 20,000 by adding pennies which have tails on both sides, then the shaking or mixing would be less effective. We should still expect as an average result to get the 100 heads but in 20,000 instead of 200. The variation will be 28 or 29 on the 100 instead of 20. And this is a better limit in such cases. Taking 28 as the limit of error on 100 instances and proportionally increasing the others so that the mean error becomes 7.8 and the probable error 5.6, we may now calculate the answer without gross mistake.
The probable variation on the 1000 deaths by accident will be 18, the mean variation will be 24.6, and the limits of variation 88.5. One such table showing in five years a mean number of deaths of about 1120 per annum gives an annual deviation of about 50 up or down of this. It will be seen at once that an improvement of 30 or 40 in any one year would be without meaning, but that an improvement of from 100 to 200 would indicate some change for the better in the circumstances of the industry. Before applying these principles to the elucidation of some of the problems of sampling it will be well to give Poisson's formula (in a modified form) and to illustrate its working.
Let x equal the number of cases of one sort, y the cases of the other sort, and z the total. In the example, z will be the 700,000 engaged in the industry; x will be the 1000 killed by accidents, and y will be the 699,000 who did not so die. The limit of deviation or error calculated by Poisson's formula will be the square root of 8xy/z. Replacing x, y and z by the figures of the example we get the square root of (8×1000×699000)/700,000, which works out to the square root of 7988.57, or 89.3. Which means that we may reasonably expect the number of deaths not to vary from 1000 by more than 89, i.e., they will be between 1090 and 910. It will be seen that this number is in very satisfactory agreement with 88.5 given by the rougher calculation based on my own experiments.
To come to the question of sampling. Consider a powder of uniform fineness and fine enough to pass through an 80 sieve. For purposes of calculation this may be assumed to be made up of particles of about one-eighth of a millimetre across (say roughly 1/200 of an inch); cubed, this gives the content as about 1/500 (strictly 1/512) of a cubic m.m. Now one cubic m.m. of water weighs 1 milligram; therefore 500 such particles if they have the specific gravity of water weigh 1 milligram, and otherwise weigh 1 milligram multiplied by the sp. gr.: 500 particles of ruby silver (Pyrargyrite)[129] will weigh 5.8 milligrams and will contain nearly 3.5 milligrams of silver.
Now suppose a portion of 3.2667 grams (1/10 Assay Ton) of silver ore to contain 500 such particles of ruby silver and no other material carrying silver: such an ore would contain 35 ozs. of silver to the ton. But the limits of variation on 500 particles would be 28[130] multiplied by the square root of 5, or 62 particles. Thus the limit of sampling error would amount to just one-eighth of the silver present, or say to rather more than 4 ozs. to the ton; the mean sampling error would be rather more than a quarter of this, or say about 1.3 ozs. to the ton.
On the other hand, if one took for the assay a charge six times greater (say about 20 grams), the number of particles would be 3000 and the limits of variation would be 28 multiplied by the square root of 30, or 153 particles, which is very closely 1/20 of the silver present, or say 1.75 ozs. to the ton, whilst the mean error would amount to about .5 ozs. to the ton.
To work these examples by Poisson's formula let us assume the gangue to have a mean sp. gr. of 3. Then 500 particles would weigh 3 milligrams; and 3.2609[131] grams would contain 543,500 particles. There would be then 500 of ruby silver and 543,500 of gangue, together 544,000, and the formula gives the square root of (8×500×543500)/544000, which works out to 63 particles as against 62 by the other method.
A practical conclusion from this is of course that either the ore must be powdered more finely or a larger portion than 3 grams must be taken for the assay. Moreover, it is evident that on such an ore no small sample must be taken containing less than several million particles.
Consider now a copper ore of the same uniform fineness containing[Pg 448] particles of copper pyrites (sp. gr. 4) of which 1000 particles will weigh 8 milligrams, mixed with gangue of which 1000 particles weigh 6 milligrams.
If one gram of such ore contain .5 gram of copper pyrites (= .1725 gram copper) and .5 gram of gangue, these will contain 62,500 and say 83,500 particles respectively. Altogether 146,000 particles. With Poisson's formula this gives the limit of sampling error as the square root of (8×62500×83500)/146000 or 521 particles. But a variation of 521 on 62,500 is a variation of .83 per cent. The percentage of copper in the ore is 17.25 per cent., and .83 per cent. of this is .14 per cent. The limits of sampling error, therefore, are 17.11 per cent. and 17.39 per cent. Again, it must be remembered that the mean sampling error would be a little over one-quarter of this, or say from 17.2 per cent. to 17.3 per cent. The practical conclusion is that a powder of this degree of fineness is not fine enough. In the last place let us consider a similar iron ore containing 90 per cent. of hæmatite (sp. gr. 5) and 10 per cent. of gangue (sp. gr. 3), 1 gram of such ore will contain 90,000 particles of hæmatite weighing .9 gram and containing .63 gram of iron with say 16,500 particles of gangue weighing .1 gram. Altogether 106,500 particles.
Poisson's formula then gives the limits of variation as the square root of (8×90000×16500)/106500 or 334 particles. But 334 on 90,000 is 0.23 on 63.0, which is the percentage of iron present. The limits of sampling error then are 62.77 per cent. and 63.23 per cent. and the mean variation is from 62.94 per cent. to 63.06 per cent.
These examples are worthy of careful consideration, and it must be remembered that the calculations are made on the assumption that the ore is made up of uniform particles of mineral of such fineness as would pass easily through an 80 sieve, but which does not pretend to represent with great exactness the fineness of the powdered ore customary in practice. They show that having passed through such a sieve is no proof of sufficient powdering, not that all ores powdered and so sifted are unfit for assaying. This last would be an absurd and illogical conclusion.
If an ore be powdered to a fairly fine sand and then be passed through a series of sieves, say a 40, 60, and 80, in such a state that little or none remains on the first, but the others retain a large proportion; then of that which comes through the 80 sieve, perhaps two-thirds by weight may be even coarser than the powder I have used in the example. Of the rest most may be of about half this diameter; the weight of the really fine powder may be quite inconsiderable. On the other hand, if the grinding be continued until, on sifting, little or nothing that is powderable remains on the sieves; then in the sifted product the proportions will be very different. This last, of course, is the only right way of powdering. Also it is evident that so much depends on the manner of powdering that nothing precise can be stated as to the average coarseness of the powder. Suppose, however, by good powdering a product is obtained which may be represented by a uniform powder with particles 1/20th of a millimetre in diameter (say roughly 1/500 inch). Compared with the previous powder, the diameter has been divided by 2.5; their number, therefore, in any given weight has been increased by the cube of 2.5, which is 15.6. But the value of a sample varies as the square root of the number of particles. Hence the reduction in size and consequent increase in number has made the sample[Pg 449] nearly four times better than before; and it will be seen that this brings the sampling error within tolerable limits.
There are one or two words of warning which should be given. In the first place, using a 90 sieve instead of an 80 must not be too much relied on; the powder I took in the example would pass through it. It is a question of good powdering rather than of fine sifting. In the second place, a set of, say half-a-dozen, assays concordant within 1 oz. where the theory gives 4 ozs. as the limit of error does not upset the theory: the theory itself states this as likely. It is the error you may get in one or two assays out of a hundred, not the error you are likely to get in any one assay, which is considered under the heading "limit of error."
Accepting the result just arrived at that a portion of 1 gram may be safely taken for an assay if the particles are 1-20th of a millimetre in diameter, the further question remains as to what weight of the original sample must be reduced to this degree of fineness. This may be answered on the principle that the same degree of excellence should be aimed at in each of a series of samplings. This principle is illustrated in the table on page 2.[** PP: page reference]
A fine sand, such as would pass a 40 sieve but be retained on a 60 sieve, would be fairly represented by particles one-quarter of a millimetre in diameter. This being five times coarser, to contain the same number of particles must be 125 times (the cube of 5) as heavy; therefore 125 grams of it can be taken with the same degree of safety as 1 gram of the finer powder. Of such a sand about this weight should be taken and reduced to the finer powder. If the ore were in coarse sand, say in particles 1 millimetre in diameter, this would be four times as coarse as that last considered, and we should have to take 64 times as much of it: 64 times 125 grams is 8 kilos, or say roughly from 15 to 20 lbs. This should be crushed to the finer size and mixed; then from 100 to 150 grams should be taken and ground to the finest powder.
There is, however, a reason why, on the coarser stuff, a smaller proportion may safely be used. This becomes more evident if we consider a still coarser sample. A heap of ore in stones about 2 inches across would be 50 times coarser than the sand, and an equivalent sample would need to be 125,000 times heavier; this would amount to about 1000 tons. Experienced samplers would say that under such conditions so large a sample was hardly necessary.
This is because I have assumed in the calculations that the grains of copper pyrites, for example, were all copper pyrites and the particles of gangue were free from copper. This would be true or nearly so for the very fine powder, but far from true in the case of the ore heap. In the heap probably few of the stones would be pure ore and still fewer would be free from copper. The stones would differ among themselves in their copper contents only within certain comparatively narrow limits. And it is evident that, if replacing one stone by another, instead of resulting in the gain or loss of all the copper one or other contained, merely affected the result to one-tenth of this amount, then a sample of 1-100th of the weight (say 10 tons) would be equally safe.
It should be remembered, however, that while the man who samples on a large scale can safely and properly reduce the size of his samples on this account, yet the principle is one which counts less and less as the stuff becomes more finely divided, and ought to be ignored in the working down of the smaller samples which come to the assayer.
[126] The 10 in 20 multiplied by 10 = 100 in 200.
[127] Multiply the errors for 100 by the square root of 10.
[128] Multiply the errors for 100 by the square root of 6990.
[129] Sp. Gr. 5.8. Silver 60 per cent.
[130] Taking 28 as the limit of variation on 100.
[131] The weight of the ore less the weight of ruby silver in it.
Acid measures, 49
Acidimetry, 323
Acidity of ores, 168
Acids, 54
strength of, 54, 75, 436
Air of mines, carbonic acid in, 428
Alkalies, 330
determination of, 331
Lawrence Smith's method for, 333, 412
separation of, 332
Alkalimetry, 323
Alkaline earths, 320
Alumina, 314
determination of, 315
in mineral phosphates, 316
separation of, 314, 316
Amalgamation, 126
Ammonia, detection of, 341
determination of, 342
in natural waters, 353
Antimony, 225
detection of, 227
dry assay for, 226
gravimetric assay, 228
separation of, 228
volumetric assay, 229
Arsenic, 381
detection of, 381
dry assay for, 382
gravimetric assay, 383
in brimstone, 393
in crude arsenic, 388, 393
in mispickel, 125, 392
iodine, assay for, 386
separation by distilling, 384
uranium acetate, assay for, 389
Volhard's method applied to, 124
Assay book, 11
note, 12
results, 7
tons, 13, 131
Assaying, 1
methods, 15
Assays, check, 154
preliminary, 147
Atomic weights, 69
table of, 433
Barium, 326
Baryta, 326
Barytes, sulphur in, 378
Base bullion, sampling of, 157
Basic acetate separation, 233
Baumé's hydrometer, 77
Beryllia, 319
Bismuth, 220
colorimetric assay, 223
detection of, 221
gravimetric determination of, 222
in commercial copper, 208
separation of, 222
Black tin, 271
an analysis of, 287
assay of, 276
copper in, 204
examination of, 285
separation by vanning, 272
Blank assays, 34
Blende, sulphur in, 375
zinc in, 266
Book, assay, 11
laboratory, 10
sample, 9
[Pg 451]Boracic acid. See Boron
Borax, examination of, 431
Boron, 429
direct determination of, 431
Brass, copper in, 194
zinc in, 265
Bromine and bromides, 361
Bronze, copper in, 194
tin in, 281
Burettes, 51
Burnt ore, silver in, 116, 118
sulphur in, 377
Cadmium, 269
gravimetric determination, 269
separation of, 269
Caesium, 339
Calcination, 22, 92, 139, 345
Calcium, 320
detection of, 321
gravimetric determination, 321
separation of, 321
titration with normal acid, 322
titration with permanganate, 322
Calculation of results, 7
Calculations from formulæ, 70
Calorific effect of coal, 419
Calorimeter, 419
Calx, 345
Carbon, 414
gravimetric determination, 416
in iron or steel, 423
Carbonates, 424
Carbonic acid in the air of mines, 428
Caustic potash = potassium hydroxide, 65
Caustic soda = sodium hydroxide, 66
Cerium, 318
Chalybite, iron in, 243
Charcoal, 21, 94
Check assays for gold, 154
for silver, 104, 113
Chlorine and chlorides, 359
Chromium, 307
gravimetric assay, 309
in chrome iron ore, 308
volumetric assay, 309
Clays, examination of, 316
Coals, 418
Cobalt, 259
detection of, 259
dry assay for, 251
gravimetric determination, 260
in hardhead, 288
separation from nickel, 442, 254, 258
Coke, 25
Common salt, examination of, 336
Concentrates, assay for gold of, 140
Colorimetric assays, 44
Copper, 175
Copper, bismuth in, 208
colorimetric assay for, 190, 203
commercial, arsenic in, 208, 388
commercial, copper in, 193
commercial, examination of, 205
cyanide assay for, 194
dry assay of, 176
dry assay, loss of, in, 176
electrolytic assay for, 190, 203
gold in, 206
iodide assay for, 199
iron in, 209, 249
lead in, 206
separation of, 183
silver in, 205
sulphur in, 207
Copper ores, solution of, 183
valuation of, 181
Copper pyrites, copper in, 179, 188, 198, 202
sulphur in, 376
Culm, 22
Cupel, 23, 142
Cupellation, loss, corrections for, 103
loss in gold, 145
loss in silver, 101
of gold lead alloys, 182
of silver lead alloys, 98, 110
temperature of, 143
Cyanicides, 169
Cyanide assay for copper, 194
for nickel, 255
for tin, 278
Cyanides, alkalinity of, 167
assay of, 167
commercial, 160
double, 161
gold-dissolving power, 162
prussic acid, 162
[Pg 452]volumetric determination of, 163, 165
Cyanide liquors, alkalinity of, 167
assay of, 164, 165
assay of, for gold, 140
assay of, for zinc, &c., 169
Daniell cells, 185
Didymium, 319
Dollars to the ton, 9
Dry assays, 16
Drying, 5, 33
Earths, 314
the alkaline, 320
Electrodes, 187
Electrolysis for copper, 184
for nickel, 254
Equations, 69
Erbia, 319
Ferrous and ferric salts, 231
Filtration, 31
Finishing point, 42
Flasks, graduated, 49
Flatting, 149
Fluorine and fluorides, 363
Fluxes, 16, 93, 136, 138, 140
Formulæ, 68
Furnaces, 25
Galena, lead in, 217, 218
Gangue, 405
iron in the, 244
Gas-measuring apparatus, 52
Gases, measurement of, 44
Gay-Lussac's assay for silver, 119
assay for silver modified, 123
German silver, copper in, 194
nickel in, 255, 259
Gold, 126
amalgamation of, 126
in cyanide liquor, 140
loss of, in cupellation, 145
loss of, in parting, 154
preparation of, 63
silver in, 157
silver in, after parting, 154
test for, 126
Gold-lead alloys, cupellation of, 142
sampling of, 158
Gold ores assay with cyanide solutions, 141
calcination of, 139
concentrates, 140
fluxing, 136, 138, 140
sampling of, 127
size of assay charges, 127
tailings, 140
Gold-parting, 150
platinum in, 145, 154, 170, 171
Gold-zinc slimes, 142
Graduated vessels, 49
Gravimetric methods, 15, 27
Halogens, 358
Hardhead, 287
an analysis of, 289
Hot plate, 30
Hydrogen, preparation of, 62
reduction by, 280
Hydrometer, 77
Ignition, 32
in hydrogen, 280
Indicators, 42
Inquartation, 146
Iodine and iodides, 362
Iridium, 171
Iron, 231
bichromate assay for, 237, 243,
carbon in, 423
colorimetric assay for, 247
ferrous and ferric, 231
gravimetric determination, 233
permanganate assay for, 236, 238
phosphorus in, 399
reduction of ferric solutions, 235, 241
separation of, 232
stannous chloride assay for, 244
volumetric assays for, 234
Iron ores, iron in, 244, 247
phosphates in, 399
Laboratory books, 9
Lanthanum, 319
[Pg 453]Lawrence Smith's method for alkalies, 333, 412
Lead, 211
colorimetric assay for, 218
detection of, 211
dry assay for, 211
gravimetric determination of, 213
in commercial copper, 206
in commercial zinc, 214
in galena, 217, 218
separation of, 211, 213
volumetric determination of, 214
Litharge, use of, in dry assays, 20, 93
Lithium, 338
Lime, 320
milk of, 321
volumetric assays for, 322
Limestone, examination of, 329
lime in, 324
Limewater, 321
Loths, 9
Magnesia, magnesium, 328
mixture, preparation of, 64
Manganese, 298
colorimetric assay, 306
detection of, 299
gravimetric determination of, 300
separation of, 299
volumetric determination of, 300
Manganese peroxide, ferrous sulphate assay for, 301
iodine assay for, 302
= manganese dioxide, 298
Manganese ore, copper in, 204
manganese in, 300
peroxide in, 302
Matte, 18
Measuring, 49
flasks, 49
gases, 44, 52
gold buttons, 133, 440,
liquids, 49
silver buttons, 106
Mechanical methods, 16
Mercury, 171
dry assay, 172
wet assay, 173
Metallic particles in ores, gold, 129
particles in ores, silver, 108
particles, tin, 278, 287
Micrometer, 133
Microscope, measuring with the, 440, 133
Mispickel, arsenic in, 125, 392
sulphur in, 376
Moisture, 7, 350
Molybdate separation for phosphates, 395
solution, preparation of, 60
Molybdenum, 311
Muffle, 25
Nessler's solution, 342
Nickel, 251
dry assay for, 251
electrolytic assay, 254
gravimetric determination of, 254
in German silver, 255, 259
separation from cobalt, 254, 258, 442
separation from iron, 258
separation from manganese, 258
separation of, 253
volumetric assay, 255
Niobium, 297
Nitre, 22
use of, in dry assays, 95
Nitrogen and nitrates, 400
Nitrometer, 403
Normal acid, normal solutions, 323
Ores, determining water in, 5, 351
drying, 5
powdering, 4, 109, 130, 448
quantities of, for an assay, 11, 27, 127
sampling, 1, 127, 444
with metallic particles, 3, 108, 129
Osmiridium, 171
Osmium, 171
Ounces to the ton, long, 107
to the ton, short, 132
Oxidation, 345
Oxides, 345
[Pg 454]determination of oxygen in, 346
Oxidising agents, 22, 95, 345
effect of nitre, 95
effect of nitric acid, 56
Oxygen, 344
equivalent, 358
in natural waters, 344, 356
in ores, 348
Palladium, 171
Parting, 150
acids, 150
in flasks, 151
in glazed crucibles, 153
in special apparatus, 156
in test tubes, 152
Phosphate, assay of apatite for, 399
assay of iron ore for, 399
Phosphates, gravimetric assay, 396
volumetric assay, 397
Phosphorus and phosphates, 394
in iron, 399
Pipette, 50, 120
Platinum, 170
in gold, 145, 154, 170
Potash, commercial examination of, 338
Potassium, 336
gravimetric determination, 337
Potassium cyanide, 22, 65, 160
commercial assay of, 167
commercial, purity of, 161
Powdering, 4, 130, 448, 109
Precipitation, 30
Precipitates, drying, 32
igniting, 32, 34
washing, 31
Preliminary assays, 104, 147
Preparation of acids, 54
of other reagents, 59
Prill, 108, 129, 278, 287
Produce, 8
Pyrarsenate of magnesia, 383
Pyrites, iron in, 244
sulphur in, 370, 376
Pyrophosphate of magnesia, 397
Quantity to be taken for an assay, 11, 27, 127
Quartation, 146
Quartering, 2
Reagents, strength of, 54
Red lead for dry assays, 20, 22, 94
Reducing agents, 21, 94
effects of charcoal, &c., 94
effect of mineral sulphides, 95, 97, 98
Reduction by hydrogen, 280
of ferric solutions, 235, 242, 244
Regulus, 18
Report form, 12
Results, calculation of, 7, 13, 16, 38, 107, 131, 132
statement of, 7
Rhodium, 171
Roasting, 22, 345
Rolling, 149
Rubidium, 340
Ruthenium, 171
Sample book, 9
Sampling, 1
effect of powdering on, 449
errors, 447
gold ores, 127
metals, 157
theory of, 444
Scorification of silver ores, 88
Scorifier, 23, 89
Selenium, 379
Separation, as sulphides, 57
basic acetate, 233
molybdate, 395
Shales, bituminous, 420
Silicon and silicates, 405
in iron, 414
Silica in rocks, 409
in slags, 414
Silicates, alkalies in, 333, 412
beryllia in, 320
examination of, 409
titanium in, 411
Silver, 87
correction for cupellation loss, 103
detection of, 87
Gay-Lussac's assay, 119
Gay-Lussac's assay modified, 123
gravimetric determination of, 117
[Pg 455]in bullion, 113
in burnt ore, 116, 118
in copper, 114, 205
in galena, 114
in lead, 113
in oxide of lead, 113
in silver precipitate, 115
loss in cupellation, 101
pure preparation of, 66
Volhard's assay, 121
volumetric methods, 119, 121, 123
Silver lead alloys, cupellation of, 98
sampling of, 157
Silver ore, crucible assay of, 90
metallic particles in, 108
scorification of, 88
Size of assay charges, 11, 27, 127
Slags, 19
Soda-lime, 425
Sodium, 334
Sodium cyanide, 160
Solution, 29
Solutions, normal, 323
standard, 36
Specific gravity, 75, 436
Speise, 19
Standard, 37
solutions, 36
Standardising, 37
Steel, carbon in, 423
chromium in, 310
manganese in, 300
Stoking, 25, 143
Strength of reagents, 54
Strontium, 324
Sulphates and sulphur, 367
gravimetric determination, 369
volumetric determination, 370
Sulphides, reducing action of, 9, 95
Sulphocyanate assay for silver, 121
Sulphur in blende, 375
in burnt ore, 377
in chalcocite, 376
in coal, 419
in copper, 207
in copper pyrites, 376
in mispickel, 376
in pyrites, 370, 376
Sulphuretted hydrogen, preparation, 57
Surcharge, 154
System in assaying, 28
Table, atomic weights, 433
comparing thermometers, 435
ounces to the long ton, 107
ounces to the short ton, 132
sp. g. ammonia, 438
sp. g. hydrochloric acid, 437
sp. g. minerals, 86
sp. g. nitric acid, 436
sp. g. sulphuric acid, 439
sp. g. water, 83
Tantalum, 297
Tartar, 20, 94
Tellurium, 379
improved test for, 150
Thallium, 219
Thorium, 317
Tin, 271 See also Black tin
assay for, by vanning, 273
copper in, 204
Cornish assay, 276
cyanide assay, 278
detection of, 279
gravimetric determination of, 281
iron in, 250
separation of, 280
volumetric assay for, 282
Tin arsenide, 284
Tin phosphide, 284
Tin slag, 290
an analysis of, 292
tin in, 290
Titanium, 292
detection of, 293
in black tin, 272, 287
in rocks, 411
separation, &c., 294
Titration, 35
indirect, 43, 72
Ton, assay, 13, 131,
long, 2240 lbs. = 32,666.6 oz., 107
short, 2000 lbs = 29,166.6 oz., 132
Tungsten, 295
Tungstic acid, 295
gravimetric determination, 296
in black tin, 285
in wolfram, 296
[Pg 456]Uranium, 312
Valuation, of copper ores, 181
Vanadium, 310
Vanning, 273
Volhard's assay applied to arsenic, 124
silver assay, 121
Volume-corrector, 53
Volumetric assay, 35, 38
Water, 7, 350
direct determination of, 351
examination of, 352
expansion of, 83
solids in, 354
Weighing, 47
small gold buttons, 131
Weights, 47
Wolfram, an analysis of, 296
tungstic acid in, 296
Yttria, 319
Zinc, 261
commercial, examination of, 268
commercial, iron in, 249
commercial, lead in, 214
dry assay, 261
gasometric assay, 266
gravimetric determination, 262
in blende, 266
in cyanide liquors, 169
in silver precipitate, 266
separation of, 262
volumetric assay, 263
Zirconia, 317
Printed by Ballantyne, Hanson & Co.
London & Edinburgh.

MESSRS. CHARLES GRIFFIN & COMPANY'S PUBLICATIONS may be obtained through any Bookseller in the United Kingdom, or will be sent Post-free on receipt of a remittance to cover published price. To prevent delay, Orders should be accompanied by a Cheque or Postal Order crossed "Union of London and Smith's Bank, Chancery Lane Branch."
*** For INDEX, see next page.

COMPLETE TECHNICAL, MEDICAL, and GENERAL CATALOGUES forwarded Post-free on Application.
LONDON:
EXETER STREET, STRAND.
Third Edition, Revised, with an Additional Chapter on Foundations. Numerous Diagrams, Examples, and Tables. Large 8vo. Cloth. 16s.
"Students of Engineering will find this Text-Book invaluable."—Architect.
"The author has certainly succeeded in producing a thoroughly practical Text-Book."—Builder.
"We can unhesitatingly recommend this work not only to the Student, as the best Text-Book on the subject, but also to the professional engineer as an exceedingly valuable book of reference."—Mechanical World.
Third Edition, Thoroughly Revised. Royal 8vo. With numerous Illustrations and 13 Lithographic Plates. Handsome Cloth. Price 3Os.
General Contents.—Part I.—Elementary Statics. Part II.—General Principles of Bridge-Construction. Part III.—The Strength of Materials. Part IV.—The Design of Bridges in Detail.
"The new edition of Mr. Fidler's work will again occupy the same conspicuous position among professional text-books and treatises as has been accorded to its predecessors. The instruction imparted is sound, simple, and full. The volume will be found valuable and useful alike to those who may wish to study only the theoretical principles enunciated, and ... to others whose object and business is ... practical."—The Engineer.
At Press. In Large 8vo. Handsome Cloth. With Copious Plates and Illustrations.
Historical and Discursive.—Dock Design.—Constructive Appliances.—Materials.—Dock and Quay Walls.—Entrance Passages and Locks.—Jetties, Wharves, and Piers.—Dock Gates and Caissons.—Transit Sheds and Warehouses.—Dock Bridges.—Graving and Repairing Docks.—Working Equipment of Docks.—Index.
*** The object of the Author has been to deal fully and comprehensively with the problems arising out of the construction and maintenance of Docks and their appanages, not simply as a record of works carried out, but as a treatise on the principles underlying their construction and an investigation of the mathematical theories involved. It is primarily intended for the student; but it is hoped that the large amount of data and material collected from various sources, and in many cases contributed specially for this book, will render it useful to the expert engineer as a work of reference; while, at the same time, of general interest to directors and others connected with the management and administration of seaports.
Third Edition. In Two Parts, Published Separately.
Principal of the Battersea Polytechnic Institute, and Head of the Engineering Department therein; formerly of the Engineering Departments of the Yorkshire College, Leeds; and Dulwich College, London.
With many Illustrations, specially prepared for the Work, and numerous Examples, for the Use of Students in Technical Schools and Colleges.
"A capital text-book, arranged on an EXCELLENT SYSTEM, calculated to give an intelligent grasp of the subject, and not the mere faculty of mechanical copying.... Mr. Wells shows how to make complete working drawings, discussing fully each step in the design."—Electrical Review.
"The first book leads easily and naturally towards the second, where the technical pupil brought into contact with large and more complex designs."—The Schoolmaster.
Third Edition, Revised and Enlarged. With additional Illustrations. Large 8vo, Handsome Cloth. 25s.
General Contents.—Gas Engines:—General Description—History and Development—British, French, and German Gas Engines—Gas Production for Motive Power—Theory of the Gas Engine—Chemical Composition of Gas in Gas Engines—Utilisation of Heat—Explosion and Combustion. Oil Motors:—History and Development—Various Types—Priestman's and other Oil Engines. Hot-Air Engines:—History and Development—Various Types: Stirling's, Ericsson's, &c., &c.
"The best book now published on Gas, Oil, and Air Engines.... Will be of very great interest to the numerous practical engineers who have to make themselves familiar with the motor of the day.... Mr. Donkin has the advantage of long practical experience, combined with high scientific and experimental knowledge, and an accurate perception of the requirements of Engineers."—The Engineer.
"We heartily recommend Mr. Donkin's work.... A monument of careful labour.... Luminous and comprehensive."—Journal of Gas Lighting.
"A thoroughly reliable and exhaustive Treatise."—Engineering.
In Quarto, Handsome Cloth. With Numerous Plates. 25s.
With many Tests and Experiments on different Types of Boilers, as to the Heating Value of Fuels, &c., with Analyses of Gases and Amount of Evaporation, and Suggestions for the Testing of Boilers.
General Contents.—Classification of different Types of Boilers—425 Experiments on English and Foreign Boilers with their Heat Efficiencies shown in Fifty Tables—Fire Grates of Various Types—Mechanical Stokers—Combustion of Fuel in Boilers—Transmission of Heat through Boiler Plates, and their Temperature—Feed Water Heaters, Superheaters, Feed Pumps, &c.—Smoke and its Prevention—Instruments used in Testing Boilers—Marine and Locomotive Boilers—Fuel Testing Stations—Discussion of the Trials and Conclusions—On the Choice of a Boiler, and Testing of Land, Marine, and Locomotive Boilers—Appendices—Bibliography—Index.
With Plates illustrating Progress made during recent years, and the best Modern Practice.
"A work of reference at present unique. Will give an answer to almost any question connected with the performance of boilers that it is possible to ask."—Engineer.
"Probably the most exhaustive résumé that has ever been collected. A practical Book by a thoroughly practical man."—Iron and Coal Trades Review.
Third Edition, Revised and Enlarged. Pocket-Size, Leather, 12s. 6d.; also Larger Size for Office Use, Cloth, 12s. 6d.
A Handbook of Rules, Formulæ, Tables, &c., relative to Material, Scantlings, and Pressures, Safety Valves, Springs, Fittings and Mountings, &c.
FOR THE USE OF ENGINEERS, SURVEYORS, BOILER-MAKERS, AND STEAM USERS.
*** To the Second and Third Editions many New Tables for Pressure, up to 200 Lbs. per Square Inch have been added.
"The most valuable work on Boilers published in England."—Shipping World.
"Contains an Enormous Quantity of Information arranged in a very convenient form.... A most useful volume ... supplying information to be had nowhere else."—The Engineer.
Third Impression. Large Crown 8vo. With numerous Illustrations. 6s.
Contents.—General Description of Marine Machinery.—The Conditions of Service and Duties of Engineers of the Royal Navy.—Entry and Conditions of Service of Engineers of the Leading S.S. Companies.—Raising Steam.—Duties of a Steaming Watch on Engines and Boilers.—Shutting off Steam.—Harbour Duties and Watches.—Adjustments and Repairs of Engines.—Preservation and Repairs of "Tank" Boilers.—The Hull and its Fittings.—Cleaning and Painting Machinery.—Reciprocating Pumps, Feed Heaters, and Automatic Feed-Water Regulators.—Evaporators.—Steam Boats.—Electric Light Machinery.—Hydraulic Machinery.—Air-Compressing Pumps.—Refrigerating Machines.—Machinery of Destroyers.—The Management of Water-Tube Boilers.—Regulations for Entry of Assistant Engineers, R.N.—Questions given in Examinations for Promotion of Engineers, R.N.—Regulations respecting Board of Trade Examinations for Engineers, &c.
"The contents cannot fail to be appreciated."—The Steamship.
"This very useful book.... Illustrations are of great importance in a work of this kind, and it is satisfactory to find that special attention has been given in this respect."—Engineers' Gazette.
In Crown 8vo, extra, with Numerous Illustrations. [Shortly.
Second Edition, Revised. With numerous Plates reduced from Working Drawings and 280 Illustrations in the Text. 21s.
Contents.—Historical Introduction, 1763-1863.—Modern Locomotives: Simple.—Modern Locomotives: Compound.—Primary Consideration in Locomotive Design.—Cylinders, Steam Chests, and Stuffing Boxes.—Pistons, Piston Rods, Crossheads, and Slide Bars.—Connecting and Coupling Rods.—Wheels and Axles, Axle Boxes, Hornblocks, and Bearing Springs.—Balancing.—Valve Gear.—Slide Valves and Valve Gear Details.—Framing, Bogies and Axle Trucks, Radial Axle Boxes.—Boilers.—Smokebox, Blast Pipe, Firebox Fittings.—Boiler Mountings.—Tenders.—Railway Brakes.—Lubrication.—Consumption of Fuel, Evaporation and Engine Efficiency.—American Locomotives.—Continental Locomotives.—Repairs, Running, Inspection, and Renewals.—Three Appendices.—Index.
"Likely to remain for many years the Standard Work for those wishing to learn Design."—Engineer.
"A most interesting and valuable addition to the bibliography of the Locomotive."—Railway Official Gazette.
"We recommend the book as thoroughly practical in its character, and meriting a place in any collection of ... works on Locomotive Engineering."—Railway News.
"The work contains all that can be learnt from a book upon such a subject. It will at once rank as the standard work upon this important subject."—Railway Magazine.
In Large 8vo. Handsome Cloth. With Plates and Illustrations. 16s.
Contents.—Discussion of the Term "Light Railways."—English Railways, Rates, and Farmers.—Light Railways in Belgium, France, Italy, other European Countries, America and the Colonies, India, Ireland.—Road Transport as an alternative.—The Light Railways Act, 1896.—The Question of Gauge.—Construction and Working.—Locomotives and Rolling-Stock.—Light Railways in England, Scotland, and Wales.—Appendices and Index.
"Mr. W.H. Cole has brought together ... a large amount of valuable information ... hitherto practically inaccessible to the ordinary reader."—Times.
"Will remain, for some time yet a Standard Work in everything relating to Light Railways."—Engineer.
"The author has extended practical experience that makes the book lucid and useful. It is exceedingly well done"—Engineering.
"The whole subject is exhaustively and practically considered. The work can be cordially recommended as indispensable to those whose duty it is to become acquainted with one of the prime necessities of the immediate future."—Railway Official Gazette.
"There could be no better book of first reference on its subject. All classes of Engineers will welcome its appearance."—Scotsman.
Third Edition, Revised and Enlarged. With Numerous Illustrations. Price 8s. 6d.
"Concise explanations illustrated by 115 very clear diagrams and drawings and 4 folding-plates ... the book fulfils a valuable function."—Athenæum.
"Mr. Hurst's valves and valve-gearing will prove a very valuable aid, and tend to the production of Engines of scientific design and economical working.... Will be largely sought after by Students and Designers.—Marine Engineer.
"Useful and thoroughly practical. Will undoubtedly be found of GREAT VALUE to all concerned with the design of Valve-gearing."—Mechanical World.
"Almost every type of valve and its gearing is clearly set forth, and illustrated in such a way as to be readily understood and practically applied by either the Engineer, Draughtsman, or Student.... Should prove both USEFUL and valuable to all Engineers seeking for reliable and clear information on the subject. Its moderate price brings it within the reach of all"—Industries and Iron.
"Mr. Hurst's work is admirably suited to the needs of the practical mechanic.... It is free from any elaborate theoretical discussions, and the explanations of the various types of valve-gear are accompanied by diagrams which render them easily understood."—The Scientific American.
Hints on Steam Engine Design and Construction. By Charles Hurst, "Author of Valves and Valve Gearing." In Paper Boards, 8vo., Cloth Back. Illustrated. Price 1s. 6d. net.
Contents.—I. Steam Pipes.—II. Valves.—III. Cylinders.—IV. Air Pumps and Condensers.—V. Motion Work.—VI. Crank Shafts and Pedestals.—VII. Valve Gear.—VIII. Lubrication.—IX. Miscellaneous Details—Index.
"A handy volume which every practical young engineer should possess."—The Model Engineer.
JUST OUT. Strongly Bound in Super Royal 8vo. Cloth Boards.
Technical Assistant to Messrs. Bryan Donkin and Clench, Ltd., and Assistant Lecturer in Mechanical Engineering at the Northampton Institute, London, E.C.
"The adoption of this system for the payment of workmen has created a demand for some handy table or series of tables, by means of which the wages may be easily found without the necessity of any calculations whatever. With the object of supplying this need, the author has compiled the following tables, which have been in practical use for some time past at a large engineering works in London, and have been found of inestimable value. Not only are they of great value as a 'time saving appliance,' the computation of the bonus or premiums earned by a number of men taking only one-tenth the time by the aid of these tables compared with ordinary calculations, but they possess the additional advantage of being less liable to error, as there is practically no possibility of a mistake occurring."—Extract from Preface.
Large 8vo, Handsome Cloth. With Illustrations, Tables, &c. 21s.
Contents.—I. Friction of Solids.—II. Liquid Friction or Viscosity, and Plastic Friction.—III. Superficial Tension.—IV. The Theory of Lubrication.—V. Lubricants, their Sources, Preparation, and Properties.—VI. Physical Properties and Methods of Examination of Lubricants.—VII. Chemical Properties and Methods of Examination of Lubricants.—VIII. The Systematic Testing of Lubricants by Physical and Chemical Methods.—IX. The Mechanical Testing of Lubricants.—X. The Design and Lubrication of Bearings.—XI. The Lubrication of Machinery.—Index.
"Destined to become a classic on the subject."—Industries and Iron.
"Contains practically all that is known on the subject. Deserves the careful attention of all Engineers."—Railway Official Guide.
Fourth Edition. Very fully Illustrated. Cloth, 4s. 6d.
General Contents.—I. Explosions caused (1) by Overheating of Plates—(2) By Defective and Overloaded Safety Valves—(3) By Corrosion, Internal or External—(4) By Defective Design and Construction (Unsupported Flue Tubes; Unstrengthened Manholes; Defective Staying; Strength of Rivetted Joints; Factor of Safety)—II. Construction of Vertical Boilers: Shells—Crown Plates and Uptake Tubes—Man-Holes, Mud-Holes, and Fire-Holes—Fireboxes—Mountings—Management—Cleaning—Table of Bursting Pressures of Steel Boilers—Table of Rivetted Joints—Specifications and Drawings of Lancashire Boiler for Working Pressures (a) 80 lbs.; (b) 200 lbs. per square inch respectively.
"A valuable companion for workmen and engineers engaged about Steam Boilers, ought to be carefully studied, and always at hand."—Coll. Guardian.
"The book is very useful, especially to steam users, artisans, and young Engineers."—Engineer.
By the same Author.
Why they Occur, and How to Prevent their Occurrence. A Practical Handbook based on Actual Experiment. With Diagram and Coloured Plate. Price 3s.
Just Out. In Crown 8vo, Handsome Cloth. With Numerous Illustrations. 5s. net.
Introduction.—Tool Grinding.—Emery Wheels.—Mounting Emery Wheels.—Emery Rings and Cylinders.—Conditions to Ensure Efficient Working.—Leading Types of Machines.—Concave and Convex Grinding.—Cup and Cone Machines.—Multiple Grinding.—"Guest" Universal and Cutter Grinding Machines.—Ward Universal Cutter Grinder.—Press.—Tool Grinding.—Lathe Centre Grinder.—Polishing.—Index.
"Deals practically with every phase of his subject."—Ironmonger.
Fifth Edition. Folio, strongly half-bound, 21/.
"Those who have experience in exact Survey-work will best know how to appreciate the enormous amount of labour represented by this valuable book. The computations enable the user to ascertain the sines and cosines for a distance of twelve miles to within half an inch, and this by Reference To But One Table, in place of the usual Fifteen minute computations required. This alone is evidence of the assistance which the Tables ensure to every user, and as every Surveyor in active practice has felt the want of such assistance few knowing of their publication will remain without them."—Engineer.
For the Use of Students preparing for Competitive Examinations. With 600 pp., over 200 Illustrations, 6 Folding Plates, and numerous Examination Papers. Thirteenth Edition, Revised. 8/6.
"Professor Jamieson fascinates the reader by his clearness of conception and simplicity of expression. His treatment recalls the lecturing of Faraday."—Athenæum.
"The Best Book yet published for the use of Students."—Engineer.
For Advanced and "Honours" Students. By Prof. Jamieson, assisted by David Robertson, B.Sc., Professor of Electrical Engineering in the Merchant Venturers' Technical College, Bristol. [Shortly.
Vol. I.—Comprising Part I.: The Principle of Work and its applications; Part II.: Gearing. Price 7s. 6d. Third Edition.
"Fully maintains the reputation of the Author."—Pract. Engineer.
Vol. II.—Comprising Parts III. to VI.: Motion and Energy; Graphic Statics; Strength of Materials; Hydraulics and Hydraulic Machinery. Second Edition. 8s. 6d.
"Well and lucidly written."—The Engineer.
*** Each of the above volumes is complete in itself, and sold separately.
Crown 8vo. With Illustrations and Examination Papers.
For First-Year Students. Ninth Edition, Revised. 3/6.
"Should be in the hands of every engineering apprentice."—Practical Engineer.
For First-Year Students. Fifth Edition.. 3/6.
"A capital text-book.... The diagrams are an important feature."—Schoolmaster.
"A thoroughly trustworthy Text-book. Practical and clear."—Nature.
Specially arranged for First-Year Students. Fifth Edition, Revised. 3/6.
"The work has very high qualities, which may be condensed into the one word 'clear.'"—Science and Art.
In Preparation. 300 pages. Crown 8vo. Profusely Illustrated.
For the Use of Electrical Engineering Students and those interested in Electric Transmission of Power.
For the Use of Electricians and Engineers. Pocket Size. Leather, 8s. 6d. Sixteenth Edition. [See p. 49.
A MANUAL OF APPLIED MECHANICS: Comprising the Principles of Statics and Cinematics, and Theory of Structures, Mechanism, and Machines. With Numerous Diagrams. Crown 8vo, cloth. Sixteenth Edition. 12s. 6d.
A MANUAL OF CIVIL ENGINEERING: Comprising Engineering Surveys, Earthwork, Foundations, Masonry, Carpentry, Metal Work, Roads, Railways, Canals, Rivers, Waterworks, Harbours, &c. With Numerous Tables and Illustrations. Crown 8vo. Cloth. Twenty-First Edition. 16s.
A MANUAL OF MACHINERY AND MILLWORK: Comprising the Geometry, Motions, Work, Strength, Construction, and Objects of Machines, &c. Illustrated with nearly 300 Woodcuts, Crown 8vo, cloth. Seventh Edition. 12s. 6d.
A MANUAL OF THE STEAM-ENGINE AND OTHER PRIME MOVERS: With a Section on Gas, Oil, and Air Engines, by Bryan Donkin, M.Inst.C.E. With Folding Plates and Numerous Illustrations. Crown 8vo, cloth. Fifteenth Edition. 12s. 6d.[Pg xxxvi]
USEFUL RULES AND TABLES: For Architects, Builders, Engineers, Founders, Mechanics, Shipbuilders, Surveyors, &c. With Appendix for the use of Electrical Engineers. By Professor Jamieson, F.R.S.E. Seventh Edition. 10s. 6d.
A MECHANICAL TEXT-BOOK: A Practical and Simple Introduction to the Study of Mechanics. By Professor Rankine and E.F. Bamber, C.E. With Numerous Illustrations. Crown 8vo, cloth. Fifth Edition. 9s.
*** The "Mechanical Text-Book" was designed by Professor Rankine as an Introduction to the above Series of Manuals.
Royal 8vo. Cloth, 31s. 6d.
Part I. Papers relating to Temperature, Elasticity, and Expansion of Vapours, Liquids, and Solids. Part II. Papers on Energy and its Transformations. Part III. Papers on Wave-Forms, Propulsion of Vessels, &c.
With Memoir by Professor Tait, M.A. Edited by W.J. Millar, C.E. With fine Portrait on Steel, Plates, and Diagrams.
"No more enduring Memorial of Professor Rankine could be devised than the publication of these papers in an accessible form.... The Collection is most valuable on account of the nature of his discoveries, and the beauty and completeness of his analysis.... The Volume exceeds in importance any work in the same department published in our time."—Architect.
THE MECHANIC'S GUIDE: A Hand-Book for Engineers and Artizans. With Copious Tables and Valuable Recipes for Practical Use. Illustrated. Second Edition. Crown 8vo. Cloth, 7/6.[Pg xxxvii]
SECOND EDITION, Revised and Enlarged. In Large 8vo, Handsome cloth, 34s.
With numerous Woodcuts, and Sixty-nine Plates.
"A Book of great Professional Usefulness."—Iron.
In Large 8vo, Handsome Cloth. With Frontispiece, several Plates, and over 250 Illustrations. 21s.
Contents—Early History of Pumping Engines—Steam Pumping Engines—Pumps and Pump Valves—General Principles of Non-Rotative Pumping Engines—The Cornish Engine, Simple and Compound—Types of Mining Engines—Pit Work—Shaft Sinking—Hydraulic Transmission of Power in Mines—Valve Gears of Pumping Engines—Water Pressure Pumping Engines—Water Works Engines—Pumping Engine Economy and Trials of Pumping Machinery—Centrifugal and other Low-Lift Pumps—Hydraulic Rams. Pumping Mains, &c.—Index.
"By the 'one' English Engineer who probably knows more about Pumping Machinery than any other.' ... A volume recording the results of long experience and study."—The Engineer.
"Undoubtedly the best and most practical treatise on Pumping Machinery that has yet been published."—Mining Journal.
Royal 8vo, Handsome Cloth. With numerous Illustrations and Tables. 25s.
In order to render the work complete for the purposes of the Shipbuilder, whether at home or abroad, the Methods of Calculation introduced by Mr. F.K. Barnes, Mr. Gray, M. Reech, M. Daymard, and Mr. Benjamin, are all given separately, illustrated by Tables and worked-out examples. The book contains more than 200 Diagrams, and is illustrated by a large number of actual cases, derived from ships of all descriptions.
"Sir Edward Reed's 'Stability of Ships' is invaluable. The Naval Architect will find brought together and ready to his hand, a mass of information which he would otherwise have to seek in an almost endless variety of publications, and some of which he would possibly not be able to obtain at all elsewhere."—Steamship.
Second Edition. Illustrated with Plates, Numerous Diagrams, and Figures in the Text. 18s. net.
Contents.—I. Manufacture of Cast Iron, Wrought Iron, and Steel.—Composition of Iron and Steel, Quality, Strength, Tests, &c. II. Classification of Steel Ships. III. Considerations in making choice of Type of Vessel.—Framing of Ships. IV. Strains experienced by Ships.—Methods of Computing and Comparing Strengths of Ships. V. Construction of Ships.—Alternative Modes of Construction.—Types of Vessels.—Turret, Self Trimming, and Trunk Steamers, &c.—Rivets and Rivetting, Workmanship. VI. Pumping Arrangements. VII. Maintenance.—Prevention of Deterioration in the Hulls of Ships.—Cement, Paint, &c.—Index.
"So thorough and well written is every chapter in the book that it is difficult to select any of them as being worthy of exceptional praise. Altogether, the work is excellent, and will prove of great value to those for whom it is intended."—The Engineer.
"Mr. Walton has written for the profession of which he is an ornament. His work will be read and appreciated, no doubt, by every M.I.N.A., and with great benefit by the majority of them."—Journal of Commerce.
Second Edition, Cloth, 8s. 6d. Leather, for the Pocket, 8s. 6d.
GRIFFIN'S ELECTRICAL PRICE-BOOK: For Electrical, Civil, Marine, and Borough Engineers, Local Authorities, Architects, Railway Contractors, &c., &c. Edited by H.J. Dowsing.
"The Electrical Price-Book removes all mystery about the cost of Electrical Power. By its aid the expense that will be entailed by utilising electricity on a large or small scale can be discovered."—Architect.
"This admirable series."—Fairplay. "A very useful series."—Nature.
"The volumes of Messrs. Griffin's Nautical Series may well and profitably be read by all interested in our national maritime progress."—Marine Engineer.
"Every Ship should have the whole Series as a Reference Library. Handsomely bound, clearly printed and illustrated."—Liverpool Journ. of Commerce.
The British Mercantile Marine: An Historical Sketch of its Rise and Development. By the Editor, Capt. Blackmore. 3s. 6d.
"Captain Blackmore's splendid book ... contains paragraphs on every point of interest to the Merchant Marine. The 243 pages of this book are the most valuable to the sea captain that have ever been compiled."—Merchant Service Review.
Elementary Seamanship. By D. Wilson-Barker, Master Mariner, F.R.S.E., F.R.G.S. With numerous Plates, two in Colours, and Frontispiece. Third Edition, Thoroughly Revised, Enlarged, and Re-set. With additional Illustrations. 6s.
"This admirable manual, by Capt. Wilson Barker, of the 'Worcester', seems to us perfectly designed. "—Athenæum.
Know Your Own Ship: A Simple Explanation of the Stability, Construction, Tonnage, and Freeboard of Ships. By Thos. Walton, Naval Architect. With numerous Illustrations and additional Chapters on Buoyancy, Trim, and Calculations. Sixth Edition, Revised. 7s. 6d.
"Mr. Walton's book will be found very useful."—The Engineer.
Navigation: Theoretical and Practical. By D. Wilson-Barker, Master Mariner, &c., and William Allingham. Second Edition, Revised. 8s. 6d.
"Precisely the kind of work required for the New Certificates of competency. Candidates will find it Invaluable."—Dundee Advertiser.
Marine Meteorology: For Officers of the Merchant Navy. By William Allingham, First Class Honours, Navigation, Science and Art Department. With Illustrations, Maps, and Diagrams, and facsimile reproduction of log page. 7s. 6d.
"Quite the best publication on this subject."—Shipping Gazette.
Latitude and Longitude: How to find them. By W.J. Millar, C.E., late Sec. to the Inst. of Engineers and Shipbuilders in Scotland. Second Edition, Revised. 2s.
"Cannot but prove an acquisition to those studying Navigation."—Marine Engineer.
Practical Mechanics: Applied to the requirements of the Sailor. By Thos. Mackenzie, Master Mariner, F.R.A.S. Second Edition, Revised. 3s. 6d.
"Well worth the money ... exceedingly helpful."—Shipping World.
Trigonometry: For the Young Sailor, &c. By Rich. C. Buck, of the Thames Nautical Training College, H.M.S. "Worcester." Second Edition, Revised. Price 3s. 6d.
"This eminently practical and reliable volume."—Schoolmaster.
Practical Algebra. By Rich. C. Buck. Companion Volume to the above, for Sailors and others. Price 3s. 6d.
"It is just the book for the young sailor mindful of progress."—Nautical Magazine.
The Legal Duties of Shipmasters. By Benedict Wm. Ginsburg, M.A., LL.D., of the Inner Temple and Northern Circuit; Barrister-at-Law. Second Edition, Thoroughly Revised and Extended. Price 4s. 6d.
"Invaluable to masters ... We can fully recommend it."—Shipping Gazette.
A Medical and Surgical Help for Shipmasters. Including First Aid at Sea. By Wm. Johnson Smith, F.R.C.S., Principal Medical Officer, Seamen's Hospital, Greenwich. Second Edition, Revised. 6s. "Sound, judicious, really helpful."—The Lancet.
Introductory Volume. Price 3s. 6d.
General Contents.—Historical: From Early Times to 1486—Progress under Henry VIII.—To Death of Mary—During Elizabeth's Reign—Up to the Reign of William III.—The 18th and 19th Centuries—Institution of Examinations—Rise and Progress of Steam Propulsion—Development of Free Trade—Shipping Legislation, 1862 to 1875—"Locksley Hall" Case—Shipmasters' Societies—Loading of Ships—Shipping Legislation, 1884 to 1894—Statistics of Shipping. The Personnel: Shipowners—Officers—Mariners—Duties and Present Position. Education: A Seaman's Education: what it should be—Present Means of Education—Hints. Discipline and Duty—Postscript—The Serious Decrease in the Number of British Seamen, a Matter demanding the Attention of the Nation.
"Interesting and Instructive ... may be read with profit and enjoyment."—Glasgow Herald.
"Every Branch of the subject is dealt with in a way which shows that the writer 'knows the ropes' familiarly."—Scotsman.
"This admirable book ... teems with useful information—Should be in the hands of every Sailor."—Western Morning News.
Third Edition, Thoroughly Revised, Enlarged, and Re-set. With Additional Illustrations. Price 6s.
With Frontispiece, Numerous Plates (Two in Colours), and Illustrations in the Text.
General Contents.—The Building of a Ship; Parts of Hull, Masts, &c.—Ropes, Knots, Splicing, &c.—Gear, Lead and Log, &c.—Rigging, Anchors—Sailmaking—The Sails, &c.—Handling of Boats under Sail—Signals and Signalling—Rule of the Road—Keeping and Relieving Watch—Points of Etiquette—Glossary of Sea Terms and Phrases—Index.
*** The volume contains the new rules of the road.
"This admirable manual, by Capt. Wilson-Barker of the 'Worcester,' seems to us perfectly designed, and holds its place excellently in Griffin's Nautical Series.' ... Although intended for those who are to become Officers of the Merchant Navy, it will be found useful by all yachtsmen."—Athenæum.
*** For complete List of Griffin's Nautical Series, see p. 39.
Second Edition, Revised and Illustrated. Price 3s. 6d.
With Numerous Illustrations and Examination Questions.
General Contents.—Definitions—Latitude and Longitude—Instruments of Navigation—Correction of Courses—Plane Sailing—Traverse Sailing—Day's Work—Parallel Sailing—Middle Latitude Sailing—Mercator's Chart—Mercator Sailing—Current Sailing—Position by Bearings—Great Circle Sailing—The Tides—Questions—Appendix: Compass Error—Numerous Useful Hints, &c.—Index.
"Precisely the kind of work required for the New Certificates of competency in grades from Second Mate to extra Master.... Candidates will find it invaluable."—Dundee Advertiser.
"A capital little book ... specially adapted to the New Examinations. The Authors are Capt. Wilson-Barker (Captain-Superintendent of the Nautical College, H.M.S. 'Worcester,' who has had great experience in the highest problems of Navigation), and Mr. Allingham, a well-known writer on the Science of Navigation and Nautical Astronomy."—Shipping World.
Handsome Cloth. Fully Illustrated. Price 7s. 6d.
With numerous Plates, Maps, Diagrams, and Illustrations, and a facsimile Reproduction of a Page from an actual Meteorological Log-Book.
Introductory.—Instruments Used at Sea for Meteorological Purposes.—Meteorological Log-Books.—Atmospheric Pressure.—Air Temperatures.—Sea Temperatures.—Winds.—Wind Force Scales.—History of the Law of Storms.—Hurricanes, Seasons, and Storm Tracks.—Solution of the Cyclone Problem.—Ocean Currents.—Icebergs.—Synchronous Charts.—Dew, Mists, Fogs, and Haze.—Clouds.—Rain, Snow, and Hail.—Mirage, Rainbows, Coronas, Halos, and Meteors.—Lightning, Corposants, and Auroras.—Questions.—Appendix.—Index.
"Quite the best publication, AND certainly the most interesting, on this subject ever presented to Nautical men."—Shipping Gazette.
*** For Complete List of Griffin's Nautical Series, see p. 39.
Second Edition, Revised. With Numerous Illustrations. Price 3s. 6d.
General Contents.—Resolution and Composition of Forces—Work done by Machines and Living Agents—The Mechanical Powers: The Lever; Derricks as Bent Levers—The Wheel and Axle: Windlass; Ship's Capstan; Crab Winch—Tackles: the "Old Man"—The Inclined Plane; the Screw—The Centre of Gravity of a Ship and Cargo—Relative Strength of Rope: Steel Wire, Manilla, Hemp, Coir—Derricks and Shears—Calculation of the Cross-breaking Strain of Fir Spar—Centre of Effort of Sails—Hydrostatics: the Diving-bell; Stability of Floating Bodies; the Ship's Pump, &c.
"This Excellent Book ... contains a LARGE AMOUNT of information."—Nature.
"Well worth the money ... will be found EXCEEDINGLY HELPFUL."—Shipping World.
"No Ships' Officers' bookcase will henceforth be complete without Captain Mackenzie's 'Practical Mechanics.' Notwithstanding my many years' experience at sea, it has told me how much more there is to acquire."—(Letter to the Publishers from a Master Mariner).
"I must express my thanks to you for the labour and care you have taken in 'Practical Mechanics.' ... It is a life's experience. ... What an amount we frequently see wasted by rigging purchases without reason and accidents to spars, &c., &c.! 'Practical Mechanics' would save all this."—(Letter to the Author from another Master Mariner).
*** Mr. Buck's Text-Book has been specially prepared with a view to the New Examinations of the Board of Trade, in which Trigonometry is an obligatory subject.
"This eminently practical and reliable volume."—Schoolmaster.
*** These elementary works on algebra and trigonometry are written specially for those who will have little opportunity of consulting a Teacher. They are books for "self-help." All but the simplest explanations have, therefore, been avoided, and answers to the Exercises are given. Any person may readily, by careful study, become master of their contents, and thus lay the foundation for a further mathematical course, if desired. It is hoped that to the younger Officers of our Mercantile Marine they will be found decidedly serviceable. The Examples and Exercises are taken from the Examination Papers set for the Cadets of the "Worcester."
"Clearly arranged, and well got up.... A first-rate Elementary Algebra."—Nautical Magazine.
Second Edition, Thoroughly Revised and Extended. In Crown 8vo. Handsome Cloth. Price 4s. 6d.
General Contents.—The Qualification for the Position of Shipmaster—The Contract with the Shipowner—The Master's Duty in respect of the Crew: Engagement; Apprentices; Discipline; Provisions, Accommodation, and Medical Comforts; Payment of Wages and Discharge—The Master's Duty in respect of the Passengers—The Master's Financial Responsibilities—The Master's Duty in respect of the Cargo—The Master's Duty in Case of Casualty—The Master's Duty to certain Public Authorities—The Master's Duty in relation to Pilots, Signals, Flags, and Light Dues—The Master's Duty upon Arrival at the Port of Discharge—Appendices relative to certain Legal Matters: Board of Trade Certificates, Dietary Scales, Stowage of Grain Cargoes, Load Line Regulations, Life-saving Appliances, Carriage of Cattle at Sea, &c., &c.—Copious Index.
"No intelligent Master should fail to add this to his list of necessary books. A few lines of it may save a lawyer's fee, besides endless worry."—Liverpool Journal of Commerce.
"Sensible, plainly written, in CLEAR and non-technical language, and will be found of much service by the Shipmaster."—British Trade Review.
Second Edition, Revised. With Diagrams. Price 2s.
"Concisely and clearly written ... cannot but prove an acquisition to those studying Navigation."—Marine Engineer.
"Young Seamen will find it handy and useful, simple and clear."—The Engineer.
*** The attention of all interested in our Merchant Navy is requested to this exceedingly useful and valuable work. It is needless to say that it is the outcome of many years practical experience amongst Seamen.
"Sound, judicious, really helpful."—The Lancet.
Sixth Edition. Revised, with Chapters on Trim, Buoyancy, and Calculations. Numerous Illustrations. Handsome Cloth, Crown 8vo. Price 7s. 6d.
This work explains, in a simple manner, such important subjects as:—
Displacement,
Deadweight,
Tonnage,
Freeboard,
Moments,
Buoyancy,
Strain,
Structure,
Stability,
Rolling,
Ballasting,
Loading,
Shifting Cargoes,
Admission of Water,
Sail Area,
&c., &c.
"The little book will be found exceedingly handy by most officers and officials connected with shipping.... Mr. Walton's work will obtain lasting success, because of its unique fitness for those for whom it has been written."—Shipping World.
"An excellent work, full of solid instruction and invaluable to every officer of the Mercantile Marine who has his profession at heart."—Shipping.
"Not one of the 242 pages could well be spared. It will admirably fulfil its purpose ... useful to ship owners, ship superintendents, ship draughtsmen, and all interested in shipping."—Liverpool Journal of Commerce.
"A mass of very useful information, accompanied by diagrams and illustrations, is given in a compact form."—Fairplay.
"We have found no one statement that we could have wished differently expressed. The matter has, so far as clearness allows, been admirably condensed, and is simple enough to be understood by every seaman."—Marine Engineer.
(See page 38.)
Fourteenth Edition, Revised. Price 21s.
Demy 8vo, Cloth. With Numerous Illustrations, reduced from Working Drawings.
General Contents.—Part I.—Principles of Marine Propulsion. Part II.—Principles of Steam Engineering. Part III.—Details of Marine Engines: Design and Calculations for Cylinders, Pistons, Valves, Expansion Valves, &c. Part IV.—Propellers. Part V.—Boilers. Part VI.—Miscellaneous.
*** This Edition includes a Chapter on Water-Tube Boilers, with Illustrations of the leading Types and the Revised Rules of the Bureau Veritas.
"In the three-fold capacity of enabling a Student to learn how to design, construct, and work a Marine Steam-Engine, Mr. Seaton's Manual has no rival."—Times.
"By far the best Manual in existence.... Gives a complete account of the methods of solving, with the utmost possible economy, the problems before the Marine Engineer."—Athenæum.
"The Student, Draughtsman, and Engineer will find this work the most valuable Handbook of Reference on the Marine Engine now in existence."—Marine Engineer.
Seventh Edition, Thoroughly Revised. Pocket-Size, Leather. 8s. 6d.
"Admirably fulfils its purpose."—Marine Engineer.
(See page 27.)
In Crown 8vo, extra, with Diagrams and Folding-Plate. 8s. 6d.
"Prof. R.H. Smith's book will be serviceable in rendering a hard road as easy as practicable for the non-mathematical Student and Engineer."—Athenæum.
"Interesting diagrams, with practical illustrations of actual occurrence, are to be found here in abundance. The very complete classified reference table will prove very useful in saving the time of those who want an integral in a hurry."—The Engineer.
Showing at a glance the Mutual Conversion of Measurements in Different Units
Of Lengths, Areas, Volumes, Weights, Stresses, Densities, Quantities of Work, Horse Powers, Temperatures, &c.
For the use of Engineers, Surveyors, Architects, and Contractors.
In 4to, Boards. 7s. 6d.
*** Prof. Smith's Conversion-Tables form the most unique and comprehensive collection ever placed before the profession. By their use much time and labour will be saved, and the chances of error in calculation diminished. It is believed that henceforth no Engineer's Office will be considered complete without them.
"The work is invaluable."—Colliery Guardian.
"Ought to be in every office where even occasional conversions are required.... Prof. Smith's Tables form very excellent checks on results."—Electrical Review.
"Prof. Smith deserves the hearty thanks, not only of the Engineer, but of the Commercial World, for having smoothed the way for the adoption of the Metric System of Measurement, a subject which is now assuming great importance as a factor in maintaining our hold upon foreign trade."—The Machinery Market.[Pg xlvii]
In Large 8vo. Handsome Cloth. 10s. 6d.
GENERAL CONTENTS.—Introduction—Chemistry of the Chief Materials of Construction—Sources of Energy—Chemistry of Steam-raising—Chemistry of Lubrication and Lubricants—Metallurgical Processes used in the Winning and Manufacture of Metals.
"The authors have succeeded beyond all expectation, and have produced a work which should give fresh power to the Engineer and Manufacturer."—The Times.
"Practical throughout ... an admirable text-book, useful not only to Students, but to Engineers and Managers of works in preventing waste and improving processes."—Scotsman.
"A book worthy to take high rank ... treatment of the subject of gaseous fuel particularly good.... Water gas and its production clearly worked out.... We warmly recommend the work."—Journal of Gas Lighting.
For Companion Volume by the same Authors, see "Chemistry for Manufacturers," p. 71.
"Clear in style and practical in method, 'The Student's Mechanics' is cordially to be commended from all points of view."—Athenæum.
Demy 8vo, with Numerous Illustrations, 9s.
General Contents.—Heat and Combustion—Fuel, Varieties of—Firing Arrangements: Furnace, Flues, Chimney—The Boiler, Choice of—Varieties—Feed-water Heaters—Steam Pipes—Water: Composition, Purification—Prevention of Scale, &c., &c.
"The Section on Heat is one of the best and most lucid ever written."—Engineer.
"Cannot fail to be valuable to thousands using steam power."—Railway Engineer.
Gas Manufacture (The Chemistry of). A Handbook on the Production, Purification, and Testing of Illuminating Gas, and the Assay of Bye-Products. By W.J.A. Butterfield, M.A., F.I.C., F.C.S. With Illustrations. Third Edition, Revised (in preparation). [See page 77.
Water Supply: A Practical Treatise on the Selection of Sources and the Distribution of Water. By Reginald E. Middleton, M. Inst. C.E., M. Inst. Mech. E., F.S.I. With Four Plates and Numerous Diagrams. Crown 8vo. [See page 77.
Central Electrical Stations: Their Design, Organisation, and Management. By C.H. Wordingham, A.K.C., M. Inst. C.E. Price 24s. net.
For details see opposite page.
Sewage Disposal Works: A Guide to the Construction of Works for the Prevention of the Pollution by Sewage of Rivers and Estuaries. By W. Santo Crimp, M. Inst. C.E., F.G.S. Second Edition, Revised and Enlarged. Large 8vo, Handsome Cloth. With 37 Plates. Price 30s. [See page 76.
Trades' Waste: Its Treatment and Utilisation, with Special Reference to the Prevention of Rivers' Pollution. By W. Naylor, F.C.S., A.M. Inst. C.E. With Numerous Plates, Diagrams, and Illustrations. 21s. net. [See page 76.
Calcareous Cements: Their Nature, Preparation, and Uses. With some Remarks upon Cement Testing. By Gilbert Redgrave, Assoc. Inst. C.E. With Illustrations, Analytical Data, and Appendices on Costs, &c. 8s. 6d. [See page 76.
Road Making and Maintenance: A Practical Treatise for Engineers, Surveyors, and others. With an Historical Sketch of Ancient and Modern Practice. By Thomas Aitken, Assoc. M. Inst. C.E., M. Assoc. Municipal and County Engrs.; M. San. Inst. With numerous Plates, Diagrams, and Illustrations. 21s. [See page 79.
Light Railways at Home and Abroad. By William Henry Cole, M. Inst. C.E., late Deputy Manager, North-Western Railway, India. Large 8vo, Handsome Cloth, Plates and illustrations. 16s. [See page 30.
Practical Sanitation: A Handbook for Sanitary Inspectors and others interested in Sanitation. By Geo. Reid, M.D., D.P.H., Medical Officer, Staffordshire County Council. With Appendix on Sanitary Law, by Herbert Manley, M.A., M.B., D.P.H. Tenth Edition, Revised. 6s. [See page 78.
Sanitary Engineering: A Practical Manual of Town Drainage and Sewage and Refuse Disposal. By Frank Wood, A.M. Inst. C.E., F.G.S., Borough Surveyor, Fulham. Fully Illustrated. 8s. 6d. net. [See page 78.
Dairy Chemistry: A Practical Handbook for Dairy Managers, Chemists, and Analysts. By H. Droop Richmond, F.C.S., Chemist to the Aylesbury Dairy Company. With Tables, Illustrations, &c. Handsome Cloth, 16s. [See page 73.
Milk: Its Production and Uses. With Chapters on Dairy Farming, The Diseases of Cattle, and on the Hygiene and Control of Supplies. By Edward F. Willoughby, M.D. (Lond.), D.P.H. (Lond. and Camb.), Inspector of Farms and General Scientific Adviser to Welford & Sons, Ltd. [See page 73.
Flesh Foods: With Methods for their Chemical, Microscopical, and Bacteriological Examination. A Handbook for Medical Men, Inspectors, Analysts, and others. By C. Ainsworth Mitchell, B.A., F.I.C., Mem. Council Soc. of Public Analysts. With numerous Illustrations and a coloured Plate. 10s. 6d. [See page 73.
Foods: Their Composition and Analysis. By A. Wynter Blyth, M.R.C.S., F.C.S., Public Analyst for the County of Devon. With Tables, Folding Plate, and Frontispiece. Fifth Edition, Thoroughly Revised. 21s. [See page 72.
"An admirable digest of the most recent state of knowledge."—Chemical News.
Introductory.—Central Station Work as a Profession.—As an Investment.—The Establishment of a Central Station.—Systems of Supply.—Site.—Architecture.—Plant.—Boilers.—Systems of Draught and Waste Heat Economy.—Coal Handling, Weighing, and Storing.—The Transmission of Steam.—Generators.—Condensing Appliances.—Switching Gear, Instruments, and Connections.—Distributing Mains.—Insulation, Resistance, and Cost.—Distributing Networks.—Service Mains and Feeders.—Testing Mains.—Meters and Appliances.—Standardising and Testing Laboratory.—Secondary Batteries.—Street Lighting.—Cost.—General Organisation.—Mains Department.—Installation Department.—Standardising Department.—Drawing Office.—Clerical Department.—The Consumer.—Routine and Main Laying.—Index.
"One of the most valuable contributions to Central Station literature we have had for some time."—Electricity.
General Principles of Switchgear Design.—Constructional Details.—Circuit Breakers or Arc Interrupting Devices.—Automatically Operated Circuit Breakers.—Alternating Reverse Current Devices.—Arrangement of 'Bus Bars, and Apparatus for Parallel Running.—General Arrangement of Controlling Apparatus for High Tension Systems.—General Arrangement of Controlling Apparatus for Low Tension Systems.—Examples of Complete Installations.—Long Distance Transmission Schemes.
Sixteenth Edition, Thoroughly Revised and Enlarged.
With Numerous Diagrams. Pocket Size. Leather, 8s. 6d.
Units of Measurement.—Measures.—Testing.—Conductors.—Dielectrics.—Submarine Cables.—Telegraphy.—Electro-Chemistry.—Electro-Metallurgy.—Batteries.—Dynamos and Motors.—Transformers.—Electric Lighting.—Miscellaneous.—Logarithms.—Appendices.
"Wonderfully Perfect.... Worthy of the highest commendation we can give it."—Electrician.
"The Sterling Value of Messrs. Munro and Jamieson's Pocket-Book."—Electrical Review.
In Five Volumes. Large 8vo. Sold Separately.
Introductory Volume, fully Illustrated. Second Edition, Revised.
Price 10s. 6d.
Contents.—Gravitation.—The Acceleration of Gravity.—Elasticity.—Stresses and Strains.—Torsion.—Bending of Rods.—Spiral Springs.—Collision.—Compressibility of Liquids.—Pressures and Volumes of Gases.—Thermal Effects Accompanying Strain.—Capillarity.—Surface Tension.—Laplace's Theory of Capillarity.—Diffusion of Liquids.—Diffusion of Gases.—Viscosity of Liquids.—Index.
Volume II. Second Edition. Fully Illustrated. Price 8s. 6d.
Contents.—The Nature of Sound and its chief Characteristics.—The Velocity of Sound in Air and other Media.—Reflection and Refraction of Sound.—Frequency and Pitch of Notes.—Resonance and Forced Oscillations.—Analysis of Vibrations.—The Transverse Vibrations of Stretched Strings or Wires.—Pipes and other Air Cavities.—Rods.—Plates.—Membranes.—Vibrations maintained by Heat.—Sensitive Flames and Jets.—Musical Sand.—The Superposition of Waves.—Index.
"The work ... may be recommended to anyone desirous of possessing an easy up-to-date Standard Treatise on Acoustics."—Literature.
"Very clearly written.... The names of the authors are a guarantee of the scientific accuracy and up-to-date character of the work."—Educational Times.
Volume III. At Press. Fully Illustrated.
Remaining Volumes in Preparation—
LIGHT; MAGNETISM AND ELECTRICITY.
THE MEAN DENSITY OF THE EARTH: An Essay to which the Adams Prize was adjudged in 1893 in the University of Cambridge. By J.H. Poynting, Sc.D., F.R.S., Late Fellow of Trinity College, Cambridge; Professor of Physics, Birmingham University. In Large 8vo, with Bibliography, Illustrations in the Text, and Seven Lithographed Plates. 12s. 6d.
"An account of this subject cannot fail to be of great and general interest to the scientific mind. Especially is this the case when the account is given by one who has contributed so considerably as has Prof. Poynting to our present state of knowledge with respect to a very difficult subject.... Remarkably has Newton's estimate been verified by Prof. Poynting."—Athenæum.[Pg li]
For Works on Chemistry and Chemical Industries see p. 69.
| PAGE | ||
| Geology, Stratigraphical, | R. Etheridge, F.R.S., | 52 |
| " Physical, | Prof. H.G. Seeley, | 52 |
| " Practical Aids, | Prof. Grenville Cole, | 53 |
| " Open Air Studies, | " " | 19 |
| Griffin's "New Land" Series, | Ed. by Prof. Cole, | 54 |
| Prospecting for Minerals, | S. Herbert Cox, A.R.S.M., | 55 |
| Food Supply, | Robt. Bruce, | 55 |
| New Lands, | H.R. Mill, D.Sc., F.R.S.E., | 54 |
| Building Construction, | Prof. James Lyon, | 54 |
| Ore and Stone Mining, | Prof. Le Neve Foster, | 56 |
| Elementary Mining, | " " | 56 |
| Coal Mining, | H.W. Hughes, F.G.S., | 56 |
| Practical Coal Mining, | G.L. Kerr, M.Inst.M.E., | 58 |
| Elementary " | " " | 58 |
| Electrical Coal Mining, | D. Burns, | 58 |
| Mine-Surveying, | Bennett H. Brough, A.R.S.M., | 57 |
| Blasting and Explosives, | O. Guttmann, A.M.I.C.E., | 57 |
| Mine Accounts, | Prof. J.G. Lawn, | 57 |
| Mining Engineers' Pkt.-Bk., | E.R. Field, M.Inst.M.M., | 60 |
| Petroleum, | Redwood and Holloway, | 61 |
| A Handbook on Petroleum, | J.H. Thomson and Dr. Redwood, | 61 |
| The Petroleum Lamp, | " " | 61 |
| Metallurgical Analysis, | Macleod and Walker, | 60 |
| Metallurgy (General), | Phillips and Bauerman, | 60 |
| " (Elementary), | Prof. Humboldt Sexton, | 66 |
| Getting Gold, | J.C.F. Johnson, F.G.S., | 58 |
| Gold Seeking in South Africa, | Theo Kassner, | 59 |
| Cyanide Process, | James Park, F.G.S., | 59 |
| Cyaniding, | Smart and Julian, | 59 |
| Electric Smelting, | Borchers and McMillan, | 67 |
| Electro-Metallurgy, | W.G. McMillan, F.I.C., | 67 |
| Assaying, | J.J. & C. Beringer, | 66 |
| Metallurgical Analysis, | J.J. Morgan, F.C.S, | 66 |
| Griffin's Metallurgical Series | Ed. by Sir W. Roberts-Austen, | 62 |
| Introduction, | Sir W. Roberts-Austen, K.C.B.,, | 63 |
| Gold, Metallurgy of, | Dr. Kirke Rose, A.R.S.M., | 63 |
| Lead and Silver, " | H.F. Collins, A.R.S.M., | 64 |
| Iron, Metallurgy of, | Thos. Turner, A.R.S.M., | 65 |
| Steel, " | F.W. Harbord, | 65 |
| Metallurgical Machinery, | H.C. Jenkins, A.R.S.M., | 64 |
| Goldsmith and Jeweller's Art, | Thos. B. Wigley, | 68 |
| Precious Stones, | Dr. Max Bauer, | 68 |
Demy 8vo, Handsome cloth, 18s.
"It is impossible to praise too highly the research which Professor Seeley's 'Physical Geology' evidences. It is far more than a Text-book—it is a Directory to the Student in prosecuting his researches."—Presidential Address to the Geological Society, 1885, by Rev. Prof. Bonney, D.Sc., LL.D., F.R.S.
"Professor Seeley maintains in his 'Physical Geology' the high reputation he already deservedly bears as a Teacher."—Dr. Henry Woodward, F.R.S., in the "Geological Magazine."
"Professor Seeley's work includes one of the most satisfactory Treatises on Lithology in the English language."—American Journal of Engineering.
Demy 8vo, Handsome cloth, 34s.
"No such compendium of geological knowledge has ever been brought together before."—Westminster Review.
"If Prof. Seeley's volume was remarkable for its originality and the breadth of its views, Mr. Etheridge fully justifies the assertion made in his preface that his book differs in construction and detail from any known manual.... Must take high rank among works of reference."—Athenæum.
For details, see Griffin's Introductory Science Series, p. 85.
Crown 8vo. Handsome Cloth, 2s. 6d.
PART I.—The Earth's Atmosphere in Remote Geological Periods.
PART II.—The Atmosphere of Our Present Period. Appendices; Index.
*** Dr. Phipson's work presents, amidst much which is of interest to the Scientist and the General Reader alike, a short résumé of his discovery of the origin of Atmospheric Oxygen, the existence of which he attributes wholly to the action of Solar Radiation upon vegetable life. The book will be found replete with much that is new, curious, and interesting, both in connection with Weather Lore, and with Scientific Meteorology.—Publisher's Note.
"The book should prove of interest to general readers, as well as to meteorologists and other students of science."—Nature.
See also the two following pages (54, 55), and page 85.
GENERAL CONTENTS.—
PART I.—Sampling of the Earth's Crust.
PART II.—Examination of Minerals.
PART III.—Examination of Rocks.
PART IV.—Examination of Fossils.
"Prof. Cole treats of the examination of minerals and rocks in a way that has never been attempted before ... deserving of the highest praise. Here indeed are 'Aids' innumerable and invaluable. All the directions are given, with the utmost clearness and precision."—Athenæum.
"That the work deserves its title, that it is full of 'Aids,' and in the highest degree 'practical,' will be the verdict of all who use it."—Nature.
"This excellent Manual ... will be a very great help.... The Section on the Examination of Fossils is probably the best of its kind yet published.... Full of well-digested information from the newest sources and from personal research."—Annals of Nat. History.
Practical Hand-Books for the Use of Prospectors, Explorers, Settlers, Colonists, and all Interested in the opening up and Development of New Lands.
In Crown 8vo. Handsome Cloth. 5s.
Introductory.—The Development of New Lands.—The Dominion of Canada.—Canada, Eastern Provinces.—Canada, Western Provinces and Territories.—Newfoundland.—The United States.—Latin America, Mexico.—Latin America, Temperate Brazil and Chili.—Latin America, Argentina.—The Falkland Islands.—Victoria.—New South Wales.—Queensland.—South Australia.—Tasmania.—Western Australia.—New Zealand.—The Resources of South Africa.—Southern Rhodesia.—Index.
"Painstaking ... complete ... of great practical assistance."—The Field.
"A want admirably supplied.... Has the advantage of being written by a professed Geographer."—Geographical Journal.
BUILDING CONSTRUCTION in WOOD, STONE, and CONCRETE. By James Lyon, M.A., Professor of Engineering in the Royal College of Science for Ireland; sometime Superintendent of the Engineering Department in the University of Cambridge; and J. Taylor, A.R.C.S.I.
*** Other Volumes, dealing with subjects of Primary Importance in the Examination and Utilisation of Lands which have not as yet been fully developed, are in preparation.[Pg lv]
Third Edition, Revised. With Illustrations. Handsome Cloth, 5s.
General Contents.—Introduction and Hints on Geology—The Determination of Minerals: Use of the Blow-pipe, &c.—Rock-forming Minerals and Non-Metallic Minerals of Commercial Value: Rock Salt, Borax, Marbles, Lithographic Stone, Quartz and Opal, &c., &c.—Precious Stones and Gems—Stratified Deposits: Coal and Ores—Mineral Veins and Lodes—Irregular Deposits—Dynamics of Lodes: Faults, &c.—Alluvial Deposits—Noble Metals: Gold, Platinum, Silver, &c.—Lead—Mercury—Copper—Tin—Zinc—Iron—Nickel, &c.—Sulphur, Antimony, Arsenic, &c.—Combustible Minerals—Petroleum—General Hints on Prospecting—Glossary—Index.
"This admirable little work ... written with scientific accuracy in a clear and lucid style.... An important addition to technical literature ... will be of value not only to the Student, but to the experienced Prospector.... If the succeeding volumes of the New Land Series are equal in merit to the First, we must congratulate the Publishers on successfully filling up a gap in existing literature.—Mining Journal.
"This excellent handbook will prove a perfect Vade-mecum to those engaged in the practical work of Mining and Metallurgy."—Times of Africa.
With many Engravings and Photographs. Handsome Cloth, 4s. 6d.
General Contents.—Climate and Soil—Drainage and Rotation of Crops—Seeds and Crops—Vegetables and Fruits—Cattle and Cattle-Breeding—Sheep and Sheep Rearing—Pigs—Poultry—Horses—The Dairy—The Farmer's Implements—The Settler's Home.
"Bristles With Information."—Farmers' Gazette.
"The work is one which will appeal to those intending to become farmers at home or in the Colonies, and who desire to obtain a general idea of the true principles of farming in all its branches."—Journal of the Royal Colonial Inst.
"A most readable and valuable book, and merits an extensive sale."—Scottish Farmer.
"Will prove of service in any part of the world."—Nature.[Pg lvi]
Fourth Edition, Revised, and brought thoroughly up-to-date by L.H. Cooke, Instructor in Mine Surveying, Royal College of Science.
INTRODUCTION. Mode of Occurrence of Minerals.—Prospecting.—Boring.—Breaking Ground.—Supporting Excavations.—Exploitation.—Haulage or Transport.—Hoisting or Winding.—Drainage.—Ventilation.—Lighting.—Descent and Ascent.—Dressing.—Principles of Employment of Mining Labour.—Legislation affecting Mines and Quarries.—Condition of the Miner.—Accidents.—Index.
"Dr. Foster's book was expected to be epoch-making, and it fully justifies such expectation.... A most admirable account of the mode of occurrence of practically all known minerals. Probably stands unrivalled for completeness."—The Mining Journal.
"This epoch-making work ... appeals to men of experience no less than to students."—Berg- und Hüttenmännische Zeitung.
"This splendid work."—Oesterr. Ztschrft. für Berg- und Hüttenwesen.
Fourth Edition, Revised and Greatly Enlarged. With Numerous Additional Illustrations, mostly reduced from Working Drawings. Price 24s. net.
Geology.—Search for Coal.—Breaking Ground.—Sinking.—Preliminary Operations.—Methods of Working.—Haulage.—Winding.—Pumping.—Ventilation.—Lighting.—Works at Surface.—Preparation of Coal for Market.—Index.
"Quite the best book of its kind ... as practical in aim as a book can be ... The illustrations are excellent."—Athenæum.
"We cordially recommend the work."—Colliery Guardian.
"Will soon come to be regarded as the standard work of its kind."—Birmingham Daily Gazette.
Tenth Edition, Revised and Enlarged. With Numerous Diagrams.
Cloth, 7s. 6d.
General Contents.—General Explanations.—Measurement of Distances.—Miner's Dial.—Variation of the Magnetic-Needle.—Surveying.—German Dial.—Theodolite.—Traversing Underground.—Surface-Surveys.—Plotting the Survey.—Calculation of Areas.—Levelling.—Measuring Distances by Telescope.—Setting-out.—Problems.—Photographic Surveying.—Appendices.
"Its clearness of style, lucidity of description, and fulness of detail have long ago won for it a place unique in the literature of this branch of mining engineering, and the present edition fully maintains the high standard of its predecessors. To the student, and to the mining engineer alike, its value is inestimable. The illustrations are excellent."—The Mining Journal.
In Large 8vo. Second Edition. Price 10s. 6d.
General Contents.—Introduction.—Part I. Engagement and Payment of Workmen.—Part II. Purchases and Sales.—Part III. Working Summaries and Analyses.—Part IV. Ledger, Balance Sheet, and Company Books.—Part V. Reports and Statistics.
"It seems impossible to suggest how Mr. Lawn's book could be made more complete or more valuable, careful, and exhaustive."—Accountants' Magazine.
In Large 8vo, with Illustrations and Folding-Plates. 10s. 6d.
"This admirable work."—Colliery Guardian.
"Should prove a vade-mecum to Mining Engineers and all engaged in practical work."—Iron and Coal Trades Review.[Pg lviii]
In Crown 8vo. Handsome cloth. With Numerous Illustrations. 6s. net.
Units of Measurement, Conductors, &c.—The Theory of the Dynamo.—The Dynamo, Details of Construction and Working.—Motors.—Lighting Installations in Collieries.—Pumping by Electricity.—Electrical Haulage.—Coal Cutting.—Miscellaneous Applications of Electricity in Mines.—Index.
"A clear and concise introduction to electrical practice in collieries."—Mining Journal.
Second Edition. Large Crown 8vo. Handsome Cloth. With over 520 Illustrations in the Text. 12s. 6d.
"An essentially practical work, and can be confidently recommended. No department of Coal-Mining has been overlooked."—Engineers' Gazette.
"This book just meets the wants of Students preparing for the Colliery Managers' Examinations. I have decided to use it for our classes here.... We have, I believe the largest atining class in Great Britain."—The Principal of a Training College.
"An abundance of information conveyed in a popular and attractive form.... Will be of great use to all who are in any way interested in coal mining."—Scottish Critic.
Second Edition. With Illustrations. Cloth, 3s. 6d.
General Contents.—Introductory: Prospecting (Alluvial and General)—Lode or Reef Prospecting—Genesiology of Gold—Auriferous Lodes—Drifts—Gold Extraction—Lixiviation—Calcination—Motor Power and its Transmission—Company Formation—Mining Appliances and Methods—Australasian Mining Regulations.
"Practical from beginning to end ... deals thoroughly with the Prospecting, Sinking, Crushing, and Extraction of gold."—Brit. Australasian.[Pg lix]
With Plates and Illustrations. Handsome Cloth. At Press.
Professor of Mining and Director of the Otago University School of Mines; late Director Thames School of Mines, and Geological Surveyor and Mining Geologist to the Government of New Zealand.
Third English Edition. Thoroughly Revised and Greatly Enlarged. With additional details concerning the Siemens-Halske and other recent processes.
Contents.—The MacArthur Process.—Chemistry of the Process.—Laboratory Experiments.—Control Testing and Analysis of Solutions.—Appliances for Cyanide Extraction.—The Actual Extraction by Cyanide.—Application of the Process.—Leaching by Agitation.—Zinc Precipitation of Gold.—The Siemens-Halske Process.—Other Cyanide Processes.—Antidotes for Cyanide Poisoning.—Cyaniding in New Zealand.
"Mr. Park's book deserves to be ranked as amongst the best of existing treatises on this subject."—Mining Journal.
At Press. With Numerous Plates, Maps, and Illustrations.
*** This book deals with the Cyanide Process from Technical, Commercial, and Scientific points of view. It is adapted for the Use of Directors, Managers, and Superintendents of Mines and Metallurgical Works, Mining Engineers, Metallurgists, Chemists, Assayers, Working Cyaniders, and Students.
In Crown. 8vo. Illustrated. Fancy Cloth Boards. 4s. 6d.
With a Chapter on the Agricultural Prospects of South Africa.
Abstract of Contents—History.—Geology.—Prospecting.—The De Kaap Goldfields.—Komati and Swaziland.—Cost of Mining, Native Labour, &c.—Lydenberg Goldfields—Zoutspanberg.—Witwatersrand.—Other Goldfields.—General Considerations—Conclusions.—Agricultural Prospects, Tables, Index, &c.
"As fascinating in its way as anything ever penned by Jules Verne. Mr. Kassner manages to impart his information in a way that enables him to be understanded even of the dullest."—African Commerce.[Pg lx]
At Press. Large 8vo. Handsome Cloth. With Illustrations.
Part I.—Qualitative Analysis and Preparation and Properties of Gases.
Part II.—Qualitative and Quantitative Analysis.
Part III.—Assaying, Technical Analysis (Gas, Water, Fuels, Oils, &c.).
*** "The aim of this work is to provide the student with a graded course of work leading from Simple Quantitative Analysis up to the Technical Quantitative Methods. It has been specially prepared to meet the requirements of Schools of Mines, and more especially, of those in the Colonies, the subject matter having been selected to cover a three years' laboratory course."—Extract from Author's Preface.
Third Edition. With Folding Plates and Many Illustrations. 36s.
General Contents.—Refractory Materials.—Fire-Clays.—Fuels, &c.—Aluminium.—Copper.—Tin.—Antimony.—Arsenic.—Zinc.—Mercury.—Bismuth.—Lead.—Iron.—Cobalt.—Nickel.—Silver.—Gold.—Platinum.
"Of the Third Edition, we are still able to say that, as a Text-book of Metallurgy, it is the best with which we are acquainted."—Engineer.
"A work which is equally valuable to the Student as a Text-book, and to the practical Smelter as a Standard Work of Reference.... The Illustrations are admirable examples of Wood Engraving."—Chemical News.
THE MINING ENGINEERS' REPORT BOOK AND DIRECTORS' AND SHAREHOLDERS' GUIDE TO MINING REPORTS. By Edwin R. Field, M.Inst.M.M. With Notes on the Valuation of Mining Property and Tabulating Reports, Useful Tables, &c., and provided with detachable blank pages for MS. Notes.
"An admirably compiled book which Mining Engineers and Managers will find extremely useful."—Mining Journal.[Pg lxi]
Second Edition. In Preparation. In Two Volumes, Large 8vo. With Numerous Maps, Plates, and Illustrations in the Text. Price 45s.
General Contents.—I. Historical.—II. Geological and Geographical Distribution of Petroleum and Natural Gas.—III. Chemical and Physical Properties.—IV. Origin—V. Production.—VI. Refining.—VII. The Shale Oil and Allied Industries.—VIII. Transport, Storage, and Distribution.—IX. Testing.—X. Application and Uses.—XI. Legislation at Home and Abroad.—XII. Statistics.—Index.
"The MOST COMPREHENSIVE AND CONVENIENT ACCOUNT that has yet appeared of a gigantic Industry which has made incalculable additions to the comfort of civilised man."—The Times.
"A splendid contribution to our technical literature."—Chemical News.
With Plates (One Coloured) and Illustrations. Price 8s. 6d. net.
Contents.—I. Introductory.—II. Sources of Supply.—III. Production.—IV. Chemical Products, Shale Oil, and Coal Tar.—V. Flash Point and Fire Test.—VI. Testings.—VII. Existing Legislation relating to Petroleum.—VIII.—IX.—Precautions Necessary.—X. Petroleum Oil Lamps.—XI. Carbide of Calcium and Acetylene.—Appendices.—Index.
"A volume that will enrich the world's petroleum literature, and render a service to the British branch of the industry.... Reliable, indispensable, a brilliant contribution."—Petroleum.
THE PETROLEUM LAMP: Its Choice and Use. A Guide to the Safe Employment of Mineral Oil in what is commonly termed the Paraffin Lamp. By Capt. J.H. Thomson and Dr. Boverton Redwood. Popular Edition, Illustrated. 1s. net.
"The book contains a great deal of interesting reading, much of which is thoroughly practical and useful. It is a work which will meet every purpose for which it has been written."—Petroleum.[Pg lxii]
In Large 8vo, Handsome Cloth. With Illustrations.
INTRODUCTION to the STUDY of METALLURGY. By the Editor. Fifth Edition. 18s. (See p. 63.)
GOLD (The Metallurgy of). By Thos. Kirke Rose, D.Sc., Assoc.R.S.M., F.I.C., Chemist and Assayer of the Royal Mint. Fourth Edition. 21s. (See p. 63.)
LEAD AND SILVER (The Metallurgy of). By H.F. Collins, Assoc.R.S.M., M.Inst.M.M. Part I., Lead, 16s; Part II., Silver, 16s. (See p. 64.)
IRON (The Metallurgy of). By T. Turner, A.R.S.M., F.I.C., F.C.S. Second Edition, Revised. 16s. (See p. 65.)
STEEL (The Metallurgy of). By F.W. Harbord, Assoc.R.S.M., F.I.C., with a Section on Mechanical Treatment by J.W. Hall, A.M.Inst, C.E. (See p. 65.) [Ready shortly.
Will be Published at Short Intervals.
METALLURGICAL MACHINERY: the Application of Engineering to Metallurgical Problems. By Henry Charles Jenkins, Wh.Sc., Assoc.R.S.M., Assoc.M.Inst.C.E., of the Royal College of Science. (See p. 64).
ALLOYS. By the Editor.
*** Other Volumes in Preparation.[Pg lxiii]
Fifth Edition, thoroughly Revised and considerably Enlarged. Large 8vo, with numerous Illustrations and Micro-Photographic Plates of different varieties of Steel. 18s.
General Contents.—The Relation of Metallurgy to Chemistry.—Physical Properties of Metals.—Alloys.—The Thermal Treatment of Metals.—Fuel and Thermal Measurements.—Materials and Products of Metallurgical Processes.—Furnaces.—Means of Supplying Air to Furnaces.—Thermo-Chemistry.—Typical Metallurgical Processes.—The Micro-Structure of Metals and Alloys.—Economic Considerations.
"No English text-book at all approaches this in the completeness with which the most modern views on the subject are dealt with. Professor Austen's volume will be invaluable, not only to the student, but also to those whose knowledge of the art is far advanced."—Chemical News.
Fourth Edition, Revised, Considerably Enlarged, and in part Re-written. Including the most recent Improvements in the Cyanide Process. With Frontispiece and numerous Illustrations. 21s.
General Contents.—The Properties of Gold and its Alloys.—Chemistry of Gold.—Mode of Occurrence and Distribution.—Placer Mining.—Shallow Deposits.—Deep Placer Mining.—Quartz Crushing in the Stamp Battery.—Amalgamation.—Other Forms of Crushing and Amalgamating.—Concentration.—Stamp Battery Practice.—Chlorination: The Preparation of Ore.—The Vat Process.—The Barrel Process.—Chlorination Practice in Particular Mills.—The Cyanide Process.—Chemistry of the Process.—Pyritic Smelting.—The Refining and Parting of Gold Bullion—The Assay of Gold Ores.—The Assay of Bullion—Economic Considerations.—Bibliography.
"A comprehensive practical treatise on this important subject."—The Times.
"The most complete description of the chlorination process which has yet been published."—Mining Journal.
"Adapted for all who are interested in the Gold Mining Industry, being free from technicalities as far as possible, but is more particularly of value to those engaged in the industry."—Cape Times.[Pg lxiv]
In Large 8vo. Handsome Cloth. With Illustrations.
In Two Volumes, Each Complete in Itself and Sold Separately.
Part I.—LEAD:
A Complete and Exhaustive Treatise on the Manufacture of Lead, with Sections on Smelting and Desilverisation, and Chapters on the Assay and Analysis of the Materials involved. Price 16s.
Summary of Contents.—Sampling and Assaying Lead and Silver.—Properties and Compounds of Lead.—Lead Ores.—Lead Smelting.—Reverberatories.—Lead Smelting in Hearths.—The Roasting of Lead Ores.—Blast Furnace Smelting; Principles, Practice, and Examples; Products.—Flue Dust, its Composition, Collection and Treatment.—Costs and Losses, Purchase of Ores.—Treatment of Zinc, Lead Sulphides, Desilverisation, Softening and Refining.—The Pattinson Process.—The Parkes Process.—Cupellation and Refining, &c., &c.
"A thoroughly sound and useful digest. May with every confidence be recommended."—Mining Journal.
Part II. SILVER.
Comprising Details regarding the Sources and Treatment of Silver Ores, together with Descriptions of Plant, Machinery, and Processes of Manufacture, Refining of Bullion, Cost of Working, &c. Price 16s.
Summary of Contents.—Properties of Silver and its Principal Compounds.—Silver Ores.—The Patio Process.—The Kazo, Fondon, Kröhnke, and Tina Processes.—The Pan Process.—Roast Amalgamation.—Treatment of Tailings and Concentration.—Retorting, Melting, and Assaying.—Chloridising-Roasting.—The Augustin, Claudet, and Ziervogel Processes.—The Hypo-Sulphite Leaching Process.—Refining.—Matte Smelting.—Pyritic Smelting.—Matte Smelting in Reverberatories.—Silver-Copper Smelting and Refining.—Index.
"The author has focussed a large amount of valuable information into a convenient form.... The author has evidently considerable practical experience, and describes the various processes clearly and well."—Mining Journal.
Ready Shortly. With Numerous Illustrations. Large 8vo. Handsome Cloth.
With over 40 Plates, 500 Illustrations (comprising nearly 100 Micro-Sections of Steel), Diagrams of Plant and Machinery, reduced from Working Drawings, and a Section on Mill Practice.
By J.W. HALL, A.M.Inst.C.E.
Abridged Contents.—The Plant, Machinery, Methods and Chemistry of the Bessemer and of the Open Hearth Processes (Acid and Basic).—The Mechanical Treatment of Steel comprising Mill Practice, Plant and Machinery.—The Influence of Metalloids, Heat Treatment, Special Steels, Microstructure, Testing, and Specifications.
Second Edition, Revised. Price 16s.
In Large 8vo, Handsome Cloth, With Numerous Illustrations (many from Photographs).
General Contents.—Early History of Iron.—Modern History of Iron.—The Age of Steel.—Chief Iron Ores.—Preparation of Iron Ores.—The Blast Furnace.—The Air used in the Blast Furnace.—Reactions of the Blast Furnace.—The Fuel used in the Blast Furnace.—Slags and Fluxes of Iron Smelting.—Properties of Cast Iron.—Foundry Practice.—Wrought Iron.—Indirect Production of Wrought Iron.—The Puddling Process.—Further Treatment of Wrought Iron.—Corrosion of Iron and Steel.
"A most valuable summary of knowledge relating to every method and stage in the manufacture of cast and wrought iron ... rich in chemical details.... Exhaustive and thoroughly up-to-date."—Bulletin of the American Iron and Steel Association.
"This is a delightful book, giving, as it does, reliable information on a subject becoming every day more elaborate."—Colliery Guardian.
"A thoroughly useful book, which brings the subject up to date. Of great value to those engaged in the iron industry."—Mining Journal.
*** For Details of Works on Mining, see pages 55-59.[Pg lxvi]
With numerous Tables and Illustrations. Crown 8vo. Cloth, 10s. 6d. Eighth Edition.
General Contents.—Part I.—Introductory; Manipulation: Sampling; Drying; Calculation of Results—Laboratory-books and Reports. Methods: Dry Gravimetric; Wet Gravimetric—Volumetric Assays: Titrometric, Colorimetric, Gasometric—Weighing and Measuring—Reagents—Formulæ, Equations, &c.—Specific Gravity.
Part II.—Metals: Detection and Assay of Silver, Gold, Platinum, Mercury, Copper, Lead, Thallium, Bismuth, Antimony, Iron, Nickel, Cobalt, Zinc, Cadmium, Tin, Tungsten, Titanium, Manganese, Chromium, &c.—Earths, Alkalies.
Part III.—Non-Metals: Oxygen and Oxides; The Halogens—Sulphur and Sulphates—Arsenic, Phosphorus, Nitrogen—Silicon, Carbon, Boron—Useful Tables.
"A really meritorious work, that may be safely depended upon either for systematic instruction or for reference."—Nature.
"This work is one of the best of its kind."—Engineer.
Third Edition, Revised. Handsome Cloth. With Numerous Illustrations. 6s.
GENERAL CONTENTS.—Introduction.—Properties of the Metals.—Combustion.—Fuels.—Refractory Materials.—Furnaces.—Occurrence of the Metals in Nature.—Preparation of the Ore for the Smelter.—Metallurgical Processes.—Iron.—Steel.—-Copper.—Lead.—Zinc and Tin.—Silver.—Gold.—Mercury.—Alloys.—Applications of Electricity to Metallurgy.—Laboratory Course.
"Just the kind of work for Students commencing the study of Metallurgy, or for Engineering Students."—Practical Engineer.
"Excellently got-up and well-arranged."—Chemical Trade Journal.
In Large 8vo. Handsome Cloth. Price 4s.
"The Author may be CONGRATULATED on the way his work has been carried out."—The Engineer.
"Will commend itself highly in Laboratory Practice. Its clearness and precision mark the book out as a highly useful one."—Mining Journal.[Pg lxvii]
Second Edition, Revised, Enlarged, and in part Re-written. With Additional Sections on Modern Theories of Electrolysis Costs, &c. Price 10s. 6d.
"This excellent treatise, ... one of the BEST and MOST COMPLETE manuals hitherto published on Electro-Metallurgy."—Electrical Review.
"This work will be a standard."—Jeweller.
"Any metallurgical process which reduces the cost of production must of necessity prove of great commercial importance.... We recommend this manual to ALL who are interested in the practical application of electrolytic processes."—Nature.
In large 8vo. With Numerous Illustrations and Three Folding-Plates. Price 21s.
Part I.—Alkalies and Alkaline Earth Metals: Magnesium, Lithium, Beryllium, Sodium, Potassium, Calcium, Strontium, Barium, the Carbides of the Alkaline Earth Metals.
Part II.—The Earth Metals: Aluminium, Cerium, Lanthanum, Didymium.
Part III.—The Heavy Metals: Copper, Silver, Gold, Zinc and Cadmium, Mercury, Tin, Lead, Bismuth, Antimony, Chromium, Molybdenum, Tungsten, Uranium, Manganese, Iron, Nickel, and Cobalt, the Platinum Group.
"Comprehensive and authoritative ... not only full of valuable information, but gives evidence of athorough insight into the technical value and possibilities of all the methods discussed."—The Electrician.
"Dr. Borchers' well-known work ... must of necessity be acquired by every one interested in the subject. Excellently put into English with additional matter by Mr. McMillan."—Nature.
"Will be of GREAT SERVICE to the practical man and the Student."—Electric Smelting.[Pg lxviii]
In Large 4to, Library Style. Beautifully Illustrated with 20 Plates, many in Colours, and 94 Figures in the Text.
General Contents.—General Properties of Gems: Their Natural Characters, Occurrence, Application, and Uses.—Detailed Description of Particular Gems: The Diamond, Rubies, Sapphires; Emeralds, Tourmalines, and Opals; Felspars, Amphiboles, Malachite.—Non-mineral Gems: Amber, &c.—Optical Features, Transparency, Translucency, Opacity, Refraction and Dispersion, &c.—Appendix: Pearls; Coral.
In Large Crown 8vo. With Numerous Illustrations. 8s. 6d.
General Contents.—Introduction.—The Ancient Goldsmith's Art.—Metallurgy of Gold.—Prices, &c.—Alloys.—Melting, Rolling, and Slitting Gold.—The workshop and Tools.—Wire Drawing.—Rings.—Chains and Insignia.—Antique Jewellery and its Revival.—Etruscan Work.—Precious Stones.—Cutting.—Polishing and Finishing.—Chasing, Embossing, and Repoussé Work.—Colouring and Finishing.—Enamelling.—Engraving.—Moulding and Casting Ornaments, &c.—Fluxes. &c.—Recovery of the Precious Metals.—Refining and Assaying.—Gilding and Electro Deposition.—Hall-Marking.—Miscellaneous.—Appendix.[Pg lxix]
For Metallurgy and Electro-Metallurgy, see previous Section.
| PAGE | ||
| Inorganic Chemistry, | Profs. Dupré and Hake, | 70 |
| Quantitative Analysis, | Prof. Humboldt Sexton, | 70 |
| Qualitative " | " " | 70 |
| Chemistry for Engineers, | Blount and Bloxam, | 71 |
| " " Manufacturers, | " " | 71 |
| Foods, Analysis of, | A. Wynter Blyth, | 72 |
| Poisons, Detection of, | " " | 72 |
| Tables for Chemists and Manufacturers, | Prof. Castell-Evans, | 79 |
| Agricultural Chemistry, | Prof. J.M.H. Munro, | 75 |
| Dairy Chemistry, | H. D. Richmond, | 73 |
| Milk, | E.F. Willoughby, | 73 |
| Flesh Foods, | C.A. Mitchell, | 73 |
| Practical Sanitation, | Dr. G. Reid, | 78 |
| Sanitary Engineering, | F. Wood, | 78 |
| Technical Mycology, | Lafar and Salter, | 74 |
| Ferments, | C. Oppenheimer, | 74 |
| Brewing, | Dr. W.J. Sykes, | 75 |
| Sewage Disposal, | Santo Crimp, | 76 |
| Trades' Waste, | W. Naylor, | 76 |
| Cements, | G.R. Redgrave, | 76 |
| Water Supply, | R.E. Middleton, | 77 |
| Road Making, | Thos. Aitken, | 79 |
| Gas Manufacture, | W. Atkinson Butterfield, | 77 |
| Acetylene, | Leeds and Butterfield, | 77 |
| Fire Risks, | Dr. Schwartz, | 77 |
| Petroleum, | Redwood and Holloway, | 61 |
| —— (Handbook), | Thomson and Redwood, | 61 |
| Oils, Soaps, Candles, | Dr. Alder Wright, | 81 |
| Lubrication and Lubricants, | Archbutt and Deeley, | 32 |
| India Rubber, | Dr. Carl O. Weber, | 81 |
| Painters' Colours, Oils, and Varnishes, | G.H. Hurst, | 80 |
| Painters' Laboratory Guide, | " " | 80 |
| Painting and Decorating, | W.J. Pearce, | 80 |
| Photography, | A. Brothers, | 21 |
| Dyeing, | Knecht and Rawson, | 82 |
| Dictionary of Dyes, | Rawson, Gardner, and Laycock, | 82 |
| Textile Printing, | Seymour Rothwell, | 83 |
| Textile Fibres of Commerce, | W.I. Hannan, | 83 |
| Dyeing and Cleaning, | G.H. Hurst, | 84 |
| Bleaching and Calico-Printing, | Geo. Duerr, | 84 |
Third Edition, Revised, Enlarged, and Re-issued. Price 6s. net.
"A well-written, clear and accurate Elementary Manual of Inorganic Chemistry.... We agree heartily with the system adopted by Drs. Dupré and Hake. Will Make Experimental Work trebly interesting because intelligible."—Saturday Review.
"There is no question that, given the perfect grounding of the Student in his Science, the remainder comes afterwards to him in a manner much more simple and easily acquired. The work is an example of the advantages of the Systematic Treatment of a Science over the fragmentary style so generally followed. By a long way the best of the small Manuals for Students."—Analyst.
With Illustrations. Fourth Edition. Crown 8vo, Cloth, 3s.
"A compact laboratory guide for beginners was wanted, and the want has been well supplied.... A good and useful book."—Lancet.
With Illustrations. Fourth Edition, Revised. Crown 8vo, Cloth, 3s. 6d.
"The work of a thoroughly practical chemist."—British Medical Journal.
"Compiled with great care, and will supply a want."—Journal of Education.
Third Edition, Revised. Crown 8vo. Cloth, 6s.
"Just the kind of work for students commencing the study of metallurgy."—Practical Engineer.[Pg lxxi]
In Two Vols., Large 8vo. With Illustrations. Sold Separately.
"The authors have succeeded beyond all expectations, and have produced a work which should give fresh power to the Engineer and Manufacturer."—The Times.
VOLUME I. Price 10s. 6d.
General Content.—INTRODUCTION—Chemistry of the Chief Materials of Construction—Sources of Energy—Chemistry of Steam-raising—Chemistry of Lubrication and Lubricants—Metallurgical Processes used in the Winning and Manufacture of Metals.
"Practical throughout ... an admirable text-book, useful not only to Students, but to Engineers and Managers of works in preventing waste and improving processes."—Scotsman.
"Eminently practical."—Glasgow Herald.
"A book worthy of high rank ... its merit is great ... treatment of the subject of gaseous fuel particularly good.... Water gas and the production clearly worked out.... Altogether a most creditable production. We warmly recommend it, and look forward with keen interest to the appearance of Vol. II."—Journal of Gas Lighting.
VOLUME II. Price 16s.
General Contents.—Sulphuric Acid Manufacture—Manufacture of Alkali, &c.—Destructive Distillation—Artificial Manure Manufacture—Petroleum—Lime and Cement—Clay Industries and Glass—Sugar and Starch—Brewing and Distilling—Oils, Resins, and Varnishes—Soap and Candles—Textiles and Bleaching—Colouring Matters, Dyeing, and Printing—Paper and Pasteboard—Pigments and Paints—Leather, Glue, and Size—Explosives and Matches—Minor Chemical Manufactures.
"Certainly a good and useful book, constituting a practical guide for students by affording a clear conception of the numerous processes as a whole."—Chemical Trade Journal.
"We confidently recommend this volume as a practical, and not overloaded, text-book, of great value to students."—The Builder.[Pg lxxii]
Just Out. Fifth Edition, Thoroughly Revised, Greatly Enlarged and Re-written. With additional Tables, Plates, and Illustrations. 21s.
General Contents.—History of Adulteration.—Legislation.—Apparatus.—"Ash."—Sugar.—Confectionery.—Honey.—Treacle.—Jams and Preserved Fruits.—Starches.—Wheaten-Flour.—Bread.—Oats.—Barley.—Rye.—Rice.—Maize.—Millet.—Potato.—Peas.—Lentils.—Beans.—Milk.—Cream.—Butter.—Oleo-Margarine.—Cheese.—Lard.—Tea.—Coffee.—Cocoa and Chocolate.—Alcohol.—Brandy.—Rum.—Whisky.—Gin.—Arrack.—Liqueurs.—Absinthe.—Yeast.—-Beer.—Wine.—Vinegar.—Lemon and Lime Juice.—Mustard.—Pepper.—Sweet and Bitter Almonds.—Annatto.—Olive Oil.—Water Analysis.—Appendix: Adulteration Acts, &c.
"Simply indispensable in the Analyst's laboratory."—The Lancet.
"A new edition of Mr. Wynter Blyth's Standard work, enriched with all the recent discoveries and improvements, will be accepted as a boon."—Chemical News.
Third Edition. In Large 8vo, Cloth, with Tables and Illustrations. Price 21s.
I.—Historical Introduction. II.—Classification—Statistics—Connection between Toxic Action and Chemical Composition—Life Tests—General Method of Procedure—The Spectroscope—Examination of Blood and Blood Stains. III.—Poisonous Gases. IV.—Acids and Alkalies. V.—More or less Volatile Poisonous Substances. VI.—Alkaloids and Poisonous Vegetable Principles. VII.—Poisons derived from Living or Dead Animal Substances. VIII.—The Oxalic Acid Group. IX.—Inorganic Poisons. Appendix: Treatment, by Antidotes or otherwise, of Cases of Poisoning.
"Undoubtedly the most complete work on Toxicology in our language."—The Analyst (on the Third Edition).
"As a practical guide, we know no better work."—The Lancet (on the Third Edition).
*** In the Third Edition, Enlarged and partly Re-written, New Analytical Methods have been introduced, and the Cadaveric Alkaloids, or Ptomaines, bodies playing so great a part in Food-poisoning and in the Manifestations of Disease, have received special attention.[Pg lxxiii]
With Numerous Tables, and 22 Illustrations. 16s.
Contents.—I. Introductory.—The Constituents of Milk. II. The Analysis of Milk. III. Normal Milk: its Adulterations and Alterations, and their Detection. IV. The Chemical Control of the Dairy. V. Biological and Sanitary Matters. VI. Butter. VII. Other Milk Products. VIII. The Milk of Mammals other than the Cow.—Appendices.—Tables.—Index.
" ... In our opinion the book is the best contribution on the subject that has yet appeared in the English language."—Lancet.
At Press, Fully Illustrated.
Crown 8vo, Handsome Cloth. Fully Illustrated. 10s. 6d.
With Numerous Tables, Illustrations, and a Coloured Plate.
Contents.—Structure and Chemical Composition of Muscular Fibre.—of Connective Tissue, and Blood.—The Flesh of Different Animals.—The Examination of Flesh.—Methods of Examining Animal Fat.—The Preservation of Flesh.—Composition and Analysis of Sausages.—Proteids of Flesh.—Meat Extracts and Flesh Peptones.—The Cooking of Flesh.—Poisonous Flesh.—The Animal Parasites of Flesh.—The Bacteriological Examination of Flesh.—The Extraction and Separation of Ptomaines.—Index.
*** This work is a complete compendium of the chemistry of animal tissues. It contains directions for the detection of morbid conditions, putrefactive changes, and poisonous or injurious constituents, together with an account of their causes and effects.—Publishers' Note.
"A compilation which will be most useful for the class for whom it is intended."—Athenæum.
"A book which no one whose duties involve considerations of food supply can afford to be without."—Municipal Journal.
In Large 8vo. Handsome Cloth. With numerous Illustrations.
Each Volume Complete in Itself, and Sold Separately.
With an Introduction by Dr. EMIL CHR. HANSEN, Principal of the Carlsberg Laboratory, Copenhagen.
Translated by CHARLES T.C. SALTER.
Vol. I.—SCHIZOMYCETIC FERMENTATION. 15s.
Including the Theory of Fermentation, the Principles of Sterilization, and Pure Culture Processes.
Vol. II., Part I. EUMYCETIC FERMENTATION. 7s. 6d.
The Morphology, Chemistry, Physiology, and Fermentative Processes of the Eumycetes, Zygomycetes, and Saccharomycetes.
"The first work of the kind which can lay claim to completeness in the treatment of a fascinating subject. The plan is admirable, the classification simple, the style is good, and the tendency of the whole volume is to convey sure information to the reader."—Lancet.
*** The publishers trust that before long they will be able to present English readers with the whole of the second volume, arrangements having been concluded whereby, upon its appearance in Germany, the English translation will be at once put in hand. This is now being done with Part I., which will be issued shortly, and which will be followed by the two final parts.
In Crown 8vo, Handsome Cloth. Price 7s. 6d. net.
Abridged Contents.—Introduction.—Definition.—Chemical Nature of Ferments.—Influence of External Factors.—Mode of Action.—Physiological Action.—Secretion.—Importance of Ferments to Vital Action.—Proteolytic Ferments.—Trypsin.—Bacteriolytic and Hæmolytic Ferments.—Vegetable Ferments.—Coagulating Ferments.—Saccharifying Ferments.—Diastases.—Polysaccharides.—Enzymes.—Ferments which decompose Glucosides.—Hydrolytic Ferments.—Lactic Acid Fermentation.—Alcoholic Fermentation.—Biology of Alcoholic Fermentation.—Oxydases.—Oxidising Fermentation.—Bibliography.—Index.
The present Translation embodies Notes and Additions to the Work made by the Author subsequent to its Publication in Germany.
"Such a veritable multum in parvo has never yet appeared. The author has set himself the task of writing a work on Ferments that should embrace human erudition on the subject"—Brewers' Journal.[Pg lxxv]
Second Edition, Revised. In Large 8vo. Handsome Cloth. With Plate and Illustrations. Price 21s.
I. Physical Principles involved in Brewing Operations.
Chemistry with special reference to the materials used in Brewing.
II. The Microscope.
Vegetable Biology.
Fermentation.
III. Water.
Barley and Malting.
Brewery Plant.
Brewing.
Beer and its Diseases.
Appendices.
Index.
"A volume of Brewing Science, which has long been awaited.... We consider it one of the most complete in contents and novel in arrangement that has yet been published.... Will command a large sale."—The Brewers' Journal.
"The appearance of a work such as this serves to remind us of the enormously rapid advances made in our knowledge of the Scientific Principles underlying the Brewing Processes.... Dr. Sykes' work will undoubtedly be of the greatest assistance, not merely to Brewers, but to all Chemists and Biologists interested in the problems which the Fermentation Industries present."—The Analyst.
"The publication of Dr. Sykes' masterly treatise on the art of Brewing is quite an event in the Brewing World.... Deserves our warmest praise.... A better guide than Dr. Sykes could hardly be found."—County Brewers' Gazette.
In Large 8vo. Handsome Cloth.
[In Preparation.[Pg lxxvi]
Second Edition, Revised and Enlarged.
With Tables, Illustrations in the Text, and 37 Lithographic Plates. Medium 8vo. Handsome Cloth. 30s.
PART I.—Introductory. PART II.—Sewage Disposal Works In Operation—Their Construction, Maintenance, and Cost.
*** From the fact of the Author's having, for some years, had charge of the Main Drainage Works of the Northern Section of the Metropolis, the chapter on London will be found to contain many important details which would not otherwise have been available.
"All persons interested in Sanitary Science owe a debt of gratitude to Mr. Crimp.... His work will be especially useful to Sanitary Authorities and their advisers ... eminently practical and useful."—Lancet.
"Probably the most complete and best treatise on the subject which has appeared in our language.... Will prove of the greatest use to all who have the problem of Sewage Disposal to face."—Edinburgh Medical Journal.
Beautifully Illustrated, with Numerous Plates, Diagrams, and Figures in the Text. 21s. net.
Contents.—I. Introduction.—II. Chemical Engineering.—III.—Wool De-greasing and Grease Recovery.—IV. Textile Industries; Calico Bleaching and Dyeing.—V. Dyeing and Calico-Printing,—VI. Tanning and Fellmongery.—VII. Brewery and Distillery Waste.—VIII. Paper Mill Refuse.—IX. General Trades' Waste.—Index.
"There is probably no person in England to-day better fitted to deal rationally with such a subject."—British Sanitarian.
"The work is thoroughly practical, and will serve as a handbook in the future for those who have to encounter the problems discussed."—Chemical Trade Journal.
In Crown 8vo, Extra. With Illustrations. 8s. 6d.
"Invaluable to the Student, Architect, and Engineer."—Building News.
"Will be useful to all interested in the manufacture, use, and testing of Cements."—Engineer.[Pg lxxvii]
Just Out. With Four Folding Plates and Numerous Illustrations. Large 8vo.
Abridged Contents.—Introductory.—Requirements as to Quality.—Requirements as to Quantity.—Storage Reservoirs.—Purification.—Service Reservoirs.—The Flow of Water through Pipes.—Distributing Systems.—Pumping Machines.—Special Requirements.
Third Edition, Revised. Fully Illustrated. At Press.
General Contents.—I. Raw Materials for Gas Manufacture.—II. Coal Gas.—III. Carburetted Water Gas.—IV. Oil Gas.—V. Enriching by Light Oils.—VI. Final Details of Manufacture.—VII. Gas Analysis.—VIII. Photometry.—IX. Applications of Gas.—X. Bye-Products.—XI. Acetylene.—Index.
"The best work of its kind which we have ever had the pleasure of reviewing."—Journal of Gas Lighting.
Just Out. With Diagrams and Illustrations. 5s. net.
General Contents.—Introductory.—Advantages of Acetylene and other Illuminants.—Chemistry and Physics.—General Principles of Acetylene Generation.—Choice of a Generator.—Statutory Regulations.—Treatment of Acetylene after Generation.—General Properties.—Mains and Service Pipes.—Subsidiary Apparatus.—Burners.—Incandescent Burners.—Heating Apparatus and Motors.—Carburetted, Compressed, and Dissolved Acetylene.—Mixtures with other Gases.—Sundry Uses.—Acetylene Lamps.—Valuation and Analysis of Carbide.
Ready Immediately. Large 8vo. Handsome Cloth. Price 16s. net.
Abridged General Contents.—Fires and Explosions of a General Character.—Dangers arising from Sources of Light and Heat.—Dangerous Gases.—Risks Attending Special Industries.—Materials Employed.—Agricultural Products.—Fats, Oils, and Resins.—Mineral Oils and Tar.—Alcohol, &c.—Metals, Oxides, Acids, &c.—Lightning, Ignition Appliances, Fireworks.[Pg lxxviii]
Tenth Edition, Revised. Price 6s.
General Contents.—Introduction—Water Supply: Drinking Water, Pollution of Water—Ventilation and Warming—Principles of Sewage Removal—Details of Drainage; Refuse Removal and Disposal—Sanitary and Insanitary Work and Appliances—Details of Plumbers' Work—House Construction—Infection and Disinfection—Food, Inspection of; Characteristics of Good Meat; Meat, Milk, Fish, &c., unfit for Human Food—Appendix: Sanitary Law; Model Bye-Laws, &c.
"Dr. Reid's very useful Manual ... abounds in practical detail."—British Medical Journal.
"A very useful Handbook, with a very useful Appendix. We recommend it not only to Sanitary Inspectors, but to Householders and all interested in Sanitary matters."—Sanitary Record.
In Crown 8vo. Handsome Cloth. Profusely Illustrated. 8s. 6d. net.
Introduction.—Hydraulics.—Velocity of Water in Pipes.—Earth Pressures and Retaining Walls.—Powers.—House Drainage.—Land Drainage.—Sewers.—Separate System.—Sewage Pumping.—Sewer Ventilation.—Drainage Areas.—Sewers, Manholes, &c.—Trade Refuse.—Sewage Disposal Works.—Bacteriolysis.—Sludge Disposal.—Construction and Cleansing of Sewers.—Refuse Disposal.—Chimneys and Foundations.
"The volume bristles with information which will be greedily read by those in need of assistance. The book is one that ought to be on the bookshelves of every practical engineer."—Sanitary Journal.
"A veritable pocket compendium of Sanitary Engineering.... A work which may, in many respects, be considered as complete ... commendably cautious ... interesting ... suggestive."—Public Health Engineer.[Pg lxxix]
Vol. I. Now Ready. in Half Morocco, 24s.
In Two Volumes, each complete in itself.
Volume I.—Chemical Engineering, Physical Chemistry. [Just Out.
Volume II.—Chemical Physics, Pure and Analytical Chemistry.
By JOHN CASTELL-EVANS, F.I.C., F.C.S., Superintendent of the Chemical Laboratories, and Lecturer on Inorganic Chemistry and Metallurgy at the Finsbury Technical College.
The Tables may almost claim to be exhaustive, and embody and collate all the most recent data established by experimentalists at home and abroad. The volumes will be found invaluable to all engaged in research and experimental investigation in Chemistry and Physics.
The Work comprehends as far as possible all rulles and tables required by the Analyst, Brewer, Distiller, Acid- and Alkali-Manufacturer, &c., &c.; and also the principal data in Thermo-chemistry, Electro-chemistry, and the various branches of Chemical Physics. Every possible care has been taken to ensure perfect accuracy, and to include the results of the most recent investigations.
In Large 8vo. Handsome Cloth. Beautifully Illustrated. With Plates and Figures in the Text. 21s.
WITH NUMEROUS PLATES, DIAGRAMS, AND ILLUSTRATIONS.
Contents.—Historical Sketch.—Resistance of Traction.—Laying out New Roads.—Earthworks, Drainage, and Retaining Walls.—Road Materials, or Metal.—Quarrying.—Stone Breaking and Haulage.—Road-Rolling and Scarifying.—The Construction of New, and the Maintenance of existing Roads.—Carriage Ways and Foot Ways.
"The Literary style is excellent ... A comprehensive and excellent Modern Book, an up-to-date work ... Should be on the reference shelf of every Municipal and County Engineer or Surveyor in the United Kingdom, and of every Colonial Engineer."—The Surveyor.[Pg lxxx]
Third Edition, Revised and Enlarged. With Illustrations, 12s. 6d.
General Contents.—Introductory—The Composition, Manufacture, Assay, and Analysis of Pigments, White, Red, Yellow and Orange, Green, Blue, Brown, and Black—Lakes—Colour and Paint Machinery—Paint Vehicles (Oils, Turpentine, &c., &c.)—Driers—Varnishes.
"A thoroughly practical book, ... the only English work that satisfactorily treats of the manufacture of oils, colours, and pigments."—Chemical Trades' Journal.
*** For Mr. Hurst's Garment Dyeing and Cleaning, see p. 84.
In Crown 8vo. Handsome Cloth. With Illustrations. 5s.
Abstract of Contents.—Preparation of Pigment Colours.—Chemical Principles Involved.—Oils and Varnishes.—Properties of Oils and Varnishes.—Tests and Experiments.—Plants, Methods, and Machinery of the Paint and Varnish Manufactures.
This Work has been designed by the Author for the Laboratory of the Technical School, and of the Paint and Colour Works, and for all interested or engaged in these industries.
Second Edition, Revised. In Crown 8vo. extra. With Numerous Illustrations and Plates (some in Colours), including Original Designs. 12s. 6d.
Introduction—Workshop and Stores—Plant and Appliances—Brushes and Tools—Materials: Pigments, Driers, Painters' Oils—Wall Hangings—Paper Hanging—Colour Mixing—Distempering—Plain Painting—Staining—Varnish and Varnishing—Imitative Painting—Graining—Marbling—Gilding—Sign-Writing and Lettering—Decoration: General Principles—Decoration in Distemper—Painted Decoration—Relievo Decoration—Colour—Measuring and Estimating—Coach-Painting—Ship-Painting.
"A thoroughly useful book ... good, sound, practical information in a clear and concise form."—Plumber and Decorator.
"A thoroughly good and reliable text-book.... So full and complete that it would be difficult to imagine how anything further could be added about the Painter's craft."—Builders' Journal.[Pg lxxxi]
In Large 8vo. Handsome Cloth. With 4 Plates and Several Illustrations. 16s. net.
Abstract of Contents.—Introduction.—The Chemistry of India Rubber.—The Examination and Valuation of India Rubber.—Examination of India Rubber Substitutes.—Inorganic Filling Materials.—Vulcanisers and Sulphur Carriers.—India Rubber Solvents.—Colouring Matters and Pigment Colours.—Constructive Components of India Rubber Articles.—Analysis of Manufactured India Rubber.—Sanitary Conditions in India Rubber Works.—Index.
"Replete with scientific and also with technical interest.... The sub-section on the physical properties is a complete résumé of every thing known to-day on the subject."—India-rubber Journal.
New Edition. In Large 8vo. Handsome Cloth. With 144 Illustrations.
"Dr. Wright's work will be found absolutely indispensable by every Chemist. Teems with information valuable alike to the Analyst and the Technical Chemist."—The Analyst.
"Will rank as the Standard English Authority on Oils and Fats for many years to come."—Industries and Iron.[Pg lxxxii]
In Two Large Volumes, 920 pp., with a Supplementary Volume, containing Specimens of Dyed Fabrics. 45s.
General Contents.—Chemical Technology of the Textile Fabrics—Water—Washing and Bleaching—Acids, Alkalies, Mordants—Natural Colouring Matters—Artificial Organic Colouring Matters—Mineral Colours—Machinery used in Dyeing—Tinctorial Properties of Colouring Matters—Analysis and Valuation of Materials used in Dyeing, &c., &c.
"The most valuable and useful work on Dyeing that has yet appeared in the English language ... likely to be the Standard Work of Reference for years to come."—Textile Mercury.
"This authoritative and exhaustive work ... the most complete we have yet seen on the subject."—Textile Manufacturer.
"The most exhaustive and complete work on the subject extant."—Textile Recorder.
Companion Volume to Knecht & Rawson's "Dyeing." In Large 8vo. Handsome Cloth, Library Style. 16s. net.
"Turn to the book as one may on any subject, or any substance in connection with the trade, and a reference is sure to be found. The authors have apparently left nothing out. Considering the immense amount of information, the book is a cheap one, and we trust it will be widely appreciated,"—Textile Mercury.[Pg lxxxiii]
In Large 8vo, Handsome Cloth, with Numerous Illustrations. 9s. net.
*** The subjects discussed in this volume are, in order to facilitate reference, arranged in alphabetical order under their respective heads. The work may thus be regarded as a Dictionary of Textile Fibres. A feature of the work is the wealth of botanical description which accompanies the Section dealing with Vegetable Fibres.—Publishers' Note.
"Useful Information.... Admirable Illustrations.... The information is not easily attainable, and in its present convenient form will be valuable."—Textile Recorder.
In Large 8vo, with Illustrations and Printed Patterns. Price 21s.
General Contents.—Introduction.—The Machinery Used in Textile Printing.—Thickeners and Mordants.—The Printing of Cotton Goods.—The Steam Style.—Colours Produced Directly on the Fibre.—Dyed Styles.—Padding Style.—Resist and Discharge Styles.—The Printing of Compound Colourings, &c.—The Printing of Woollen Goods.—The Printing of Silk Goods.—Practical Recipes for Printing.—Appendix.—Useful Tables.—Patterns.
"By far the best and most practical book on textile printing which has yet been brought out, and will long remain the standard work on the subject. It is essentially practical in character."—Textile Mercury.
"The most practical manual of textile printing which has yet appeared. We have no hesitation in recommending it."—The Textile Manufacturer.[Pg lxxxiv]
Large 8vo. Handsome Cloth. 12s. 6d.
With Illustrations and upwards of One Hundred Dyed and Printed Patterns designed specially to show various Stages of the Processes described.
GENERAL CONTENTS.—Cotton, Composition of; Bleaching, New Processes; Printing, Hand-Block; Flat-Press Work; Machine Printing—Mordants—Styles of Calico-Printing: The Dyed or Madder Style, Resist Padded Style, Discharge and Extract Style, Chromed or Raised Colours, Insoluble Colours, &c.—Thickeners—Natural Organic Colouring Matters—Tannin Matters—Oils, Soaps, Solvents—Organic Acids—Salts—Mineral Colours—Coal Tar Colours—Dyeing—Water, Softening of—Theory of Colours—Weights and Measures, &c.
"When a ready way out of a difficulty is wanted, it is in books like this that it is found."—Textile Recorder.
"Mr. Duerr's work will be found most useful.... The information given is of great value.... The Recipes are thoroughly practical."—Textile Manufacturer.
Second Edition. Revised and Enlarged. With Numerous Illustrations. 4s. 6d.
General Contents.—Technology of the Textile Fibres—Garment Cleaning—Dyeing of Textile Fabrics—Bleaching—Finishing of Dyed and Cleaned Fabrics—Scouring and Dyeing of Skin Rugs and Mats—Cleaning and Dyeing of Feathers—Glove Cleaning and Dyeing—Straw Bleaching and Dyeing—Glossary of Drugs and Chemicals—Useful Tables.
"An up-to-date hand book has long been wanted, and Mr. Hurst has done nothing more complete than this. An important work, the more so that several of the branches of the craft here treated upon are almost entirely without English Manuals for the guidance of workers. The price brings it within the reach of all."—Dyer and Calico-Printer.
"Mr. Hurst's work decidedly fills a want ... ought to be in the hands of every GARMENT DYER and cleaner in the Kingdom"—Textile Mercury.[Pg lxxxv]
"Boys could not have a more alluring introduction to scientific pursuits than these charming-looking volumes."—Letter to the Publishers from the Headmaster of one of our great Public Schools.
Handsome Cloth, 7s. 6d. Gilt, for Presentation, 8s. 6d.
General Contents.—A Daisy-Starred Pasture—Under the Hawthorns—By the River—Along the Shingle—A Fragrant Hedgerow—A Connemara Bog—Where the Samphire grows—A Flowery Meadow—Among the Corn (a Study in Weeds)—In the Home of the Alpines—A City Rubbish-Heap—Glossary.
"A fresh and stimulating book ... should take a high place.... The Illustrations are drawn with much skill."—The Times.
"Beautifully illustrated.... One of the most accurate as well as interesting books of the kind we have seen."—Athenæum.
"Redolent with the scent of woodland and meadow."—The Standard.
With 12 Full-Page Illustrations from Photographs. Cloth. Second Edition, Revised. 8s. 6d.
General Contents.—The Materials of the Earth—A Mountain Hollow—Down the Valley—Along the Shore—Across the Plains—Dead Volcanoes—A Granite Highland—The Annals of the Earth—The Surrey Hills—The Folds of the Mountains.
"The fascinating 'Open-Air Studies' of Prof. Cole give the Subject a glow animation ... cannot fail to arouse keen interest in geology."—Geological Magazine.
"A charming book, beautifully illustrated."—Athenæum.
Beautifully Illustrated. With a Frontispiece in Colours, and Numerous Specially Drawn Plates by Charles Whymper. 7s. 6d.
The Spacious Air.—The Open Fields and Downs.—In the Hedgerows.—On Open Heath and Moor.—On the Mountains.—Amongst the Evergreens.—Copse and Woodland.—By Stream and Pool.—The Sandy Wastes and Mud-flats.—Sea-laved Rocks.—Birds of the Cities.—Index.
"Enriched with excellent illustrations. A welcome addition to all libraries."—Westminster Review.[Pg lxxxvi]
Twentieth Annual Issue. Handsome cloth, 7s. 6d. (To Subscribers, 6s.).
Comprising (together with other Official Information) LISTS of the PAPERS read during the Session 1902-1903 before all the LEADING SOCIETIES throughout the Kingdom engaged in the following Departments of Research:—
§ 1. Science Generally: i.e., Societies occupying themselves with several Branches of Science, or with Science and Literature jointly.
§ 2. Mathematics and Physics.
§ 3. Chemistry and Photography.
§ 4. Geology, Geography, and Mineralogy.
§ 5. Biology, including Microscopy and Anthropology.
§ 6. Economic Science and Statistics.
§ 7. Mechanical Science, Engineering, and Architecture.
§ 8. Naval and Military Science.
§ 9. Agriculture and Horticulture.
§ 10. Law.
§ 11. Literature.
§ 12. Psychology.
§ 13. Archæology.
§ 14. Medicine.
"Fills a very real want."—Engineering.
"Indispensable to any one who may wish to keep himself abreast of the scientific work of the day."—Edinburgh Medical Journal.
"The Year-book of Societies is a Record which ought to be of the greatest use for the progress of Science."—Lord Playfair, F.R.S., K.C.B., M.P., Past-President of the British Association.
"It goes almost without saying that a Handbook of this subject will be in time one of the most generally useful works for the library or the desk."—The Times.
"British Societies are now well represented in the 'Year-Book of the Scientific and Learned Societies of Great Britain and Ireland'"—(Art. "Societies" in New Edition of "Encyclopædia Britannica," vol. xxii.)
Copies of the First Issue, giving an Account of the History, Organization, and Conditions of Membership of the various Societies, and forming the groundwork of the Series, may still be had, price 7/6. Also Copies of the Issues following.
The year-book of societies forms a complete index to the scientific work of the sessional year in the various Departments. It is used as a Handbook in all our great Scientific Centres, Museums, and Libraries throughout the Kingdom, and has become an indispensable book of reference to every one engaged in Scientific Work.
Fifth Edition, Thoroughly Revised and Considerably Enlarged. 18s.
INTRODUCTION TO THE STUDY OF METALLURGY. By Sir W.C. Roberts-Austen. With additional Illustrations and Micro-Photographic Plates of Different Varieties of Steel.
Fourth Edition, Thoroughly Revised and Enlarged. 21s. THE METALLURGY OF GOLD. By T. Kirke Rose, D.Sc., Assayer of the Royal Mint. Including the most recent Improvements in the Cyanide Process. With new Frontispiece and additional Illustrations.
In Two Volumes, each complete in itself.
THE METALLURGY OF LEAD AND SILVER. By H.F. Collins, Assoc. R.S.M., M.Inst.M.M. Part I.—LEAD. With Sections on Smelting and Desilverisation, and the Assay and Analysis of the Materials Involved. 16s. Part II.—SILVER. Sources and Treatment of Ores, with Descriptions of Plant, Machinery, &c. 16s.
Second Edition, Revised. With numerous Illustrations. 16s. THE METALLURGY OF IRON. By Thomas Turner, F.I.C., Assoc. R.S.M. Professor of Metallurgy at the University of Birmingham.
Large 8vo. Profusely Illustrated with Plates and Diagrams. THE METALLURGY OF STEEL. By F.W. Harbord. With a Section on the Mechanical Treatment of Steel. By J.W. Hall. 25s. net.
ASSAYING. By J.J. Beringer, F.I.C., F.C.S., and C. Beringer, F.C.S. Ninth Edition, Revised. With Diagrams, 10s. 6d.
GETTING GOLD. A Practical Treatise for Prospectors and Miners. By J.C.F. Johnson, A.I.M.E.. F.G.S., Life Member Australian Mine Managers' Association. Numerous Illustrations. Second Edition. 3s. 6d.
GOLD SEEKING IN SOUTH AFRICA. A Handbook of Hints for Intending Explorers, Prospectors, and Settlers. By Theo. Kassner. With a Chapter on Agriculture. Crown 8vo, Fancy Cloth Boards. Illustrated. 4s. 6d. net.
THE CYANIDE PROCESS OF GOLD EXTRACTION. By James Park, F.G.S., M.Inst.M.M., late Geological Surveyor and Mining Geologist to the New Zealand Government. New Edition, Revised and Enlarged from the last New Zealand Edition. With Frontispiece, Plates and Illustrations. 6s. net.
PROSPECTING FOR MINERALS. A Practical Handbook. By S. Herbert Cox, Assoc. R.S.M., M.Inst.M.M., F.G.S., &c. With Illustrations. Second Edition, Revised. Cloth, 5s.; Leather, 6s. 6d.
TABLES FOR QUANTITATIVE METALLURGICAL ANALYSIS FOR LABORATORY USE. By J. James Morgan, F.C.S. Large 8vo, strongly bound, cloth, 4s.
ELECTRIC SMELTING AND REFINING. By Dr. W. Borchers. Translated by Walter G. McMillan, F.I.C., F.C.S., from the Second German Edition. With numerous Illustrations and Three Folding Plates. 21s.
MINE ACCOUNTS AND MINING BOOKKEEPING. From the Actual Practice of leading Mining Companies. By James G. Lawn, Assoc. R.S.M., A.M.Inst.C.E.F.G.S. Edited by Sir Le Neve Foster, D.Sc., F.R.S. Large 8vo. Third Edition. 10s. 6d.
Large 8vo. Handsome Cloth. Illustrated, 12s. 6d. net. METALLURGICAL ANALYSIS AND ASSAYING: A Three Years' Course for Students. By W.A. Macleod, B.A., B.Sc., A.O.S.M. (N.Z.), and Chas. Walker, F.C.S.
Part I.—Qualitative Analysis, and Preparation and Properties of Cases. Part II.—Qualitative and Quantitative Analysis. Part III.—Assaying, Technical Analysis (Gas, Water, Fuels, Oils, &c.).
LONDON: CHARLES GRIFFIN & CO., Ltd., Exeter Street, Strand.
***END OF THE PROJECT GUTENBERG EBOOK A TEXTBOOK OF ASSAYING: FOR THE USE OF THOSE CONNECTED WITH MINES.***
******* This file should be named 18751-h.txt or 18751-h.zip *******
This and all associated files of various formats will be found in:
http://www.gutenberg.org/1/8/7/5/18751
Updated editions will replace the previous one--the old editions will be renamed.
Creating the works from public domain print editions means that no one owns a United States copyright in these works, so the Foundation (and you!) can copy and distribute it in the United States without permission and without paying copyright royalties. Special rules, set forth in the General Terms of Use part of this license, apply to copying and distributing Project Gutenberg-tm electronic works to protect the PROJECT GUTENBERG-tm concept and trademark. Project Gutenberg is a registered trademark, and may not be used if you charge for the eBooks, unless you receive specific permission. If you do not charge anything for copies of this eBook, complying with the rules is very easy. You may use this eBook for nearly any purpose such as creation of derivative works, reports, performances and research. They may be modified and printed and given away--you may do practically ANYTHING with public domain eBooks. Redistribution is subject to the trademark license, especially commercial redistribution.
*** START: FULL LICENSE ***
THE FULL PROJECT GUTENBERG LICENSE
PLEASE READ THIS BEFORE YOU DISTRIBUTE OR USE THIS WORK
To protect the Project Gutenberg-tm mission of promoting the free
distribution of electronic works, by using or distributing this work
(or any other work associated in any way with the phrase "Project
Gutenberg"), you agree to comply with all the terms of the Full Project
Gutenberg-tm License (available with this file or online at
http://www.gutenberg.org/license).
Section 1. General Terms of Use and Redistributing Project Gutenberg-tm
electronic works
1.A. By reading or using any part of this Project Gutenberg-tm
electronic work, you indicate that you have read, understand, agree to
and accept all the terms of this license and intellectual property
(trademark/copyright) agreement. If you do not agree to abide by all
the terms of this agreement, you must cease using and return or destroy
all copies of Project Gutenberg-tm electronic works in your possession.
If you paid a fee for obtaining a copy of or access to a Project
Gutenberg-tm electronic work and you do not agree to be bound by the
terms of this agreement, you may obtain a refund from the person or
entity to whom you paid the fee as set forth in paragraph 1.E.8.
1.B. "Project Gutenberg" is a registered trademark. It may only be
used on or associated in any way with an electronic work by people who
agree to be bound by the terms of this agreement. There are a few
things that you can do with most Project Gutenberg-tm electronic works
even without complying with the full terms of this agreement. See
paragraph 1.C below. There are a lot of things you can do with Project
Gutenberg-tm electronic works if you follow the terms of this agreement
and help preserve free future access to Project Gutenberg-tm electronic
works. See paragraph 1.E below.
1.C. The Project Gutenberg Literary Archive Foundation ("the Foundation"
or PGLAF), owns a compilation copyright in the collection of Project
Gutenberg-tm electronic works. Nearly all the individual works in the
collection are in the public domain in the United States. If an
individual work is in the public domain in the United States and you are
located in the United States, we do not claim a right to prevent you from
copying, distributing, performing, displaying or creating derivative
works based on the work as long as all references to Project Gutenberg
are removed. Of course, we hope that you will support the Project
Gutenberg-tm mission of promoting free access to electronic works by
freely sharing Project Gutenberg-tm works in compliance with the terms of
this agreement for keeping the Project Gutenberg-tm name associated with
the work. You can easily comply with the terms of this agreement by
keeping this work in the same format with its attached full Project
Gutenberg-tm License when you share it without charge with others.
1.D. The copyright laws of the place where you are located also govern
what you can do with this work. Copyright laws in most countries are in
a constant state of change. If you are outside the United States, check
the laws of your country in addition to the terms of this agreement
before downloading, copying, displaying, performing, distributing or
creating derivative works based on this work or any other Project
Gutenberg-tm work. The Foundation makes no representations concerning
the copyright status of any work in any country outside the United
States.
1.E. Unless you have removed all references to Project Gutenberg:
1.E.1. The following sentence, with active links to, or other immediate
access to, the full Project Gutenberg-tm License must appear prominently
whenever any copy of a Project Gutenberg-tm work (any work on which the
phrase "Project Gutenberg" appears, or with which the phrase "Project
Gutenberg" is associated) is accessed, displayed, performed, viewed,
copied or distributed:
This eBook is for the use of anyone anywhere at no cost and with
almost no restrictions whatsoever. You may copy it, give it away or
re-use it under the terms of the Project Gutenberg License included
with this eBook or online at www.gutenberg.org
1.E.2. If an individual Project Gutenberg-tm electronic work is derived
from the public domain (does not contain a notice indicating that it is
posted with permission of the copyright holder), the work can be copied
and distributed to anyone in the United States without paying any fees
or charges. If you are redistributing or providing access to a work
with the phrase "Project Gutenberg" associated with or appearing on the
work, you must comply either with the requirements of paragraphs 1.E.1
through 1.E.7 or obtain permission for the use of the work and the
Project Gutenberg-tm trademark as set forth in paragraphs 1.E.8 or
1.E.9.
1.E.3. If an individual Project Gutenberg-tm electronic work is posted
with the permission of the copyright holder, your use and distribution
must comply with both paragraphs 1.E.1 through 1.E.7 and any additional
terms imposed by the copyright holder. Additional terms will be linked
to the Project Gutenberg-tm License for all works posted with the
permission of the copyright holder found at the beginning of this work.
1.E.4. Do not unlink or detach or remove the full Project Gutenberg-tm
License terms from this work, or any files containing a part of this
work or any other work associated with Project Gutenberg-tm.
1.E.5. Do not copy, display, perform, distribute or redistribute this
electronic work, or any part of this electronic work, without
prominently displaying the sentence set forth in paragraph 1.E.1 with
active links or immediate access to the full terms of the Project
Gutenberg-tm License.
1.E.6. You may convert to and distribute this work in any binary,
compressed, marked up, nonproprietary or proprietary form, including any
word processing or hypertext form. However, if you provide access to or
distribute copies of a Project Gutenberg-tm work in a format other than
"Plain Vanilla ASCII" or other format used in the official version
posted on the official Project Gutenberg-tm web site (www.gutenberg.org),
you must, at no additional cost, fee or expense to the user, provide a
copy, a means of exporting a copy, or a means of obtaining a copy upon
request, of the work in its original "Plain Vanilla ASCII" or other
form. Any alternate format must include the full Project Gutenberg-tm
License as specified in paragraph 1.E.1.
1.E.7. Do not charge a fee for access to, viewing, displaying,
performing, copying or distributing any Project Gutenberg-tm works
unless you comply with paragraph 1.E.8 or 1.E.9.
1.E.8. You may charge a reasonable fee for copies of or providing
access to or distributing Project Gutenberg-tm electronic works provided
that
- You pay a royalty fee of 20% of the gross profits you derive from
the use of Project Gutenberg-tm works calculated using the method
you already use to calculate your applicable taxes. The fee is
owed to the owner of the Project Gutenberg-tm trademark, but he
has agreed to donate royalties under this paragraph to the
Project Gutenberg Literary Archive Foundation. Royalty payments
must be paid within 60 days following each date on which you
prepare (or are legally required to prepare) your periodic tax
returns. Royalty payments should be clearly marked as such and
sent to the Project Gutenberg Literary Archive Foundation at the
address specified in Section 4, "Information about donations to
the Project Gutenberg Literary Archive Foundation."
- You provide a full refund of any money paid by a user who notifies
you in writing (or by e-mail) within 30 days of receipt that s/he
does not agree to the terms of the full Project Gutenberg-tm
License. You must require such a user to return or
destroy all copies of the works possessed in a physical medium
and discontinue all use of and all access to other copies of
Project Gutenberg-tm works.
- You provide, in accordance with paragraph 1.F.3, a full refund of any
money paid for a work or a replacement copy, if a defect in the
electronic work is discovered and reported to you within 90 days
of receipt of the work.
- You comply with all other terms of this agreement for free
distribution of Project Gutenberg-tm works.
1.E.9. If you wish to charge a fee or distribute a Project Gutenberg-tm
electronic work or group of works on different terms than are set
forth in this agreement, you must obtain permission in writing from
both the Project Gutenberg Literary Archive Foundation and Michael
Hart, the owner of the Project Gutenberg-tm trademark. Contact the
Foundation as set forth in Section 3 below.
1.F.
1.F.1. Project Gutenberg volunteers and employees expend considerable
effort to identify, do copyright research on, transcribe and proofread
public domain works in creating the Project Gutenberg-tm
collection. Despite these efforts, Project Gutenberg-tm electronic
works, and the medium on which they may be stored, may contain
"Defects," such as, but not limited to, incomplete, inaccurate or
corrupt data, transcription errors, a copyright or other intellectual
property infringement, a defective or damaged disk or other medium, a
computer virus, or computer codes that damage or cannot be read by
your equipment.
1.F.2. LIMITED WARRANTY, DISCLAIMER OF DAMAGES - Except for the "Right
of Replacement or Refund" described in paragraph 1.F.3, the Project
Gutenberg Literary Archive Foundation, the owner of the Project
Gutenberg-tm trademark, and any other party distributing a Project
Gutenberg-tm electronic work under this agreement, disclaim all
liability to you for damages, costs and expenses, including legal
fees. YOU AGREE THAT YOU HAVE NO REMEDIES FOR NEGLIGENCE, STRICT
LIABILITY, BREACH OF WARRANTY OR BREACH OF CONTRACT EXCEPT THOSE
PROVIDED IN PARAGRAPH F3. YOU AGREE THAT THE FOUNDATION, THE
TRADEMARK OWNER, AND ANY DISTRIBUTOR UNDER THIS AGREEMENT WILL NOT BE
LIABLE TO YOU FOR ACTUAL, DIRECT, INDIRECT, CONSEQUENTIAL, PUNITIVE OR
INCIDENTAL DAMAGES EVEN IF YOU GIVE NOTICE OF THE POSSIBILITY OF SUCH
DAMAGE.
1.F.3. LIMITED RIGHT OF REPLACEMENT OR REFUND - If you discover a
defect in this electronic work within 90 days of receiving it, you can
receive a refund of the money (if any) you paid for it by sending a
written explanation to the person you received the work from. If you
received the work on a physical medium, you must return the medium with
your written explanation. The person or entity that provided you with
the defective work may elect to provide a replacement copy in lieu of a
refund. If you received the work electronically, the person or entity
providing it to you may choose to give you a second opportunity to
receive the work electronically in lieu of a refund. If the second copy
is also defective, you may demand a refund in writing without further
opportunities to fix the problem.
1.F.4. Except for the limited right of replacement or refund set forth
in paragraph 1.F.3, this work is provided to you 'AS-IS,' WITH NO OTHER
WARRANTIES OF ANY KIND, EXPRESS OR IMPLIED, INCLUDING BUT NOT LIMITED TO
WARRANTIES OF MERCHANTIBILITY OR FITNESS FOR ANY PURPOSE.
1.F.5. Some states do not allow disclaimers of certain implied
warranties or the exclusion or limitation of certain types of damages.
If any disclaimer or limitation set forth in this agreement violates the
law of the state applicable to this agreement, the agreement shall be
interpreted to make the maximum disclaimer or limitation permitted by
the applicable state law. The invalidity or unenforceability of any
provision of this agreement shall not void the remaining provisions.
1.F.6. INDEMNITY - You agree to indemnify and hold the Foundation, the
trademark owner, any agent or employee of the Foundation, anyone
providing copies of Project Gutenberg-tm electronic works in accordance
with this agreement, and any volunteers associated with the production,
promotion and distribution of Project Gutenberg-tm electronic works,
harmless from all liability, costs and expenses, including legal fees,
that arise directly or indirectly from any of the following which you do
or cause to occur: (a) distribution of this or any Project Gutenberg-tm
work, (b) alteration, modification, or additions or deletions to any
Project Gutenberg-tm work, and (c) any Defect you cause.
Section 2. Information about the Mission of Project Gutenberg-tm
Project Gutenberg-tm is synonymous with the free distribution of
electronic works in formats readable by the widest variety of computers
including obsolete, old, middle-aged and new computers. It exists
because of the efforts of hundreds of volunteers and donations from
people in all walks of life.
Volunteers and financial support to provide volunteers with the
assistance they need, is critical to reaching Project Gutenberg-tm's
goals and ensuring that the Project Gutenberg-tm collection will
remain freely available for generations to come. In 2001, the Project
Gutenberg Literary Archive Foundation was created to provide a secure
and permanent future for Project Gutenberg-tm and future generations.
To learn more about the Project Gutenberg Literary Archive Foundation
and how your efforts and donations can help, see Sections 3 and 4
and the Foundation web page at http://www.gutenberg.org/fundraising/pglaf.
Section 3. Information about the Project Gutenberg Literary Archive
Foundation
The Project Gutenberg Literary Archive Foundation is a non profit
501(c)(3) educational corporation organized under the laws of the
state of Mississippi and granted tax exempt status by the Internal
Revenue Service. The Foundation's EIN or federal tax identification
number is 64-6221541. Contributions to the Project Gutenberg
Literary Archive Foundation are tax deductible to the full extent
permitted by U.S. federal laws and your state's laws.
The Foundation's principal office is located at 4557 Melan Dr. S.
Fairbanks, AK, 99712., but its volunteers and employees are scattered
throughout numerous locations. Its business office is located at
809 North 1500 West, Salt Lake City, UT 84116, (801) 596-1887, email
business@pglaf.org. Email contact links and up to date contact
information can be found at the Foundation's web site and official
page at http://www.gutenberg.org/about/contact
For additional contact information:
Dr. Gregory B. Newby
Chief Executive and Director
gbnewby@pglaf.org
Section 4. Information about Donations to the Project Gutenberg
Literary Archive Foundation
Project Gutenberg-tm depends upon and cannot survive without wide
spread public support and donations to carry out its mission of
increasing the number of public domain and licensed works that can be
freely distributed in machine readable form accessible by the widest
array of equipment including outdated equipment. Many small donations
($1 to $5,000) are particularly important to maintaining tax exempt
status with the IRS.
The Foundation is committed to complying with the laws regulating
charities and charitable donations in all 50 states of the United
States. Compliance requirements are not uniform and it takes a
considerable effort, much paperwork and many fees to meet and keep up
with these requirements. We do not solicit donations in locations
where we have not received written confirmation of compliance. To
SEND DONATIONS or determine the status of compliance for any
particular state visit http://www.gutenberg.org/fundraising/pglaf
While we cannot and do not solicit contributions from states where we
have not met the solicitation requirements, we know of no prohibition
against accepting unsolicited donations from donors in such states who
approach us with offers to donate.
International donations are gratefully accepted, but we cannot make
any statements concerning tax treatment of donations received from
outside the United States. U.S. laws alone swamp our small staff.
Please check the Project Gutenberg Web pages for current donation
methods and addresses. Donations are accepted in a number of other
ways including checks, online payments and credit card donations.
To donate, please visit: http://www.gutenberg.org/fundraising/donate
Section 5. General Information About Project Gutenberg-tm electronic
works.
Professor Michael S. Hart is the originator of the Project Gutenberg-tm
concept of a library of electronic works that could be freely shared
with anyone. For thirty years, he produced and distributed Project
Gutenberg-tm eBooks with only a loose network of volunteer support.
Project Gutenberg-tm eBooks are often created from several printed
editions, all of which are confirmed as Public Domain in the U.S.
unless a copyright notice is included. Thus, we do not necessarily
keep eBooks in compliance with any particular paper edition.
Each eBook is in a subdirectory of the same number as the eBook's
eBook number, often in several formats including plain vanilla ASCII,
compressed (zipped), HTML and others.
Corrected EDITIONS of our eBooks replace the old file and take over
the old filename and etext number. The replaced older file is renamed.
VERSIONS based on separate sources are treated as new eBooks receiving
new filenames and etext numbers.
Most people start at our Web site which has the main PG search facility:
http://www.gutenberg.org
This Web site includes information about Project Gutenberg-tm,
including how to make donations to the Project Gutenberg Literary
Archive Foundation, how to help produce our new eBooks, and how to
subscribe to our email newsletter to hear about new eBooks.
EBooks posted prior to November 2003, with eBook numbers BELOW #10000,
are filed in directories based on their release date. If you want to
download any of these eBooks directly, rather than using the regular
search system you may utilize the following addresses and just
download by the etext year.
http://www.gutenberg.org/dirs/etext06/
(Or /etext 05, 04, 03, 02, 01, 00, 99,
98, 97, 96, 95, 94, 93, 92, 92, 91 or 90)
EBooks posted since November 2003, with etext numbers OVER #10000, are
filed in a different way. The year of a release date is no longer part
of the directory path. The path is based on the etext number (which is
identical to the filename). The path to the file is made up of single
digits corresponding to all but the last digit in the filename. For
example an eBook of filename 10234 would be found at:
http://www.gutenberg.org/dirs/1/0/2/3/10234
or filename 24689 would be found at:
http://www.gutenberg.org/dirs/2/4/6/8/24689
An alternative method of locating eBooks:
http://www.gutenberg.org/dirs/GUTINDEX.ALL
*** END: FULL LICENSE ***