
Each book in crown 8vo, cloth, with many illustrations, charts, etc., 2/6 net
OTHERS IN PREPARATION

Copyright by Messrs Flatters & Garnett, Manchester
BACTERIA NODULES ON THE ROOT OF LUPIN
PITMAN’S COMMON COMMODITIES AND INDUSTRIES
BY
G. H. J. ADLAM,
M.A., B.Sc., F.C.S.
Editor of “The School Science Review”
London
Sir Isaac Pitman & Sons, Ltd., 1 Amen Corner, E.C.4
Bath, Melbourne and New York
Printed by Sir Isaac Pitman & Sons, Ltd., London, Bath, Melbourne and New York
It has often been said, and still more often implied, that considerations of utility in education are incompatible with its main object, which is the training of the mind. Extremely divergent views have been expressed on this point. Schoolmen have looked askance at some branches of knowledge because they were supposed to be tainted with the possibility of usefulness in after life. On the other hand, business men and others have complained bitterly of the present state of education because very little that is considered “useful” has up to the present been taught in schools.
It is possible to err in both directions. A university professor, lecturing on higher Mathematics, is reported to have told his audience that it was a source of great satisfaction to him that the theorem which he was demonstrating could never be applied to anything “useful.” On the other hand, we have the well-authenticated story of the man who took his son to the Royal School of Mines to “learn copper,” and not to waste his time over other parts of Chemistry, because “they would be of no use to him.”
For narrowness of outlook, there is nothing to choose between the pedant and the “practical” man. National education would deteriorate if its control should ever pass into the hands of extremists of either type, for nothing worthy of the name of education could ever be given or received in such an irrational spirit.
In dealing with the subject of “Acids, Alkalis, and Salts,” I have endeavoured to give prominence to the commercial and domestic importance of the substances dealt with. I thereby hope to gain the interest of the vi reader, since interest stands in the same relation to education that petrol does to the motor-car. It is not education itself, but it is the source of its motive power. I have also included some considerations of a theoretical nature which may well be taken as a first step towards the continuation of the study of Chemistry.
My sincere thanks are offered to my colleagues, F. W. G. Foat, M.A., D.Litt., and Mr. I. S. Scarf, F.I.C., for much valuable help and advice; to Sir Edward Thorpe, C.B., F.R.S., and Messrs. William Collins & Sons for permission to reproduce Figures 3, 11, and 14; to Messrs. Longmans & Co. for Figures 4, 5, 9, 12, 13, 16; Messrs. Macmillan & Co., for Figures 8, 10 and 15. I have also availed myself of the assistance of several standard works on Chemistry. My acknowledgments in this direction take the practical form of the short bibliography which follows—
G. H. J. A.
Acids. A vague hint from Nature gave mankind the first indication of the existence of acids. The juice pressed from ripe grapes is a sweetish liquid. If it is kept for some time, the sweetness goes, and the liquid acquires a burning taste. If kept still longer, the burning taste is lost, and in its place a sharp acid flavour, not entirely displeasing to the palate, is developed. The liquid obtained in this way is now called wine vinegar; the particular substance which gives it its characteristic taste is acetic acid.
The strongest vinegar does not contain more than 10 per cent. of acetic acid, which is itself a comparatively weak acid. It is, therefore, not a very active solvent. Nevertheless, for metals and for limestone rock, and other substances of a calcareous nature, its solvent power is greater than that of any other liquid known at the time of its discovery. It was this property which seems to have appealed most strongly to the imagination of the early chemists; and, as is very often the case, the description of its powers was very much exaggerated. Livy and Plutarch, who have given us an account of Hannibal’s invasion of Italy by way of the Alps, both gravely declare that the 2 Carthaginian leader cleared a passage for his elephants through solid rocks by pouring vinegar over them!
In the Middle Ages, the study of Chemistry was fostered mainly as a possible means whereby long life and untold riches might be obtained. The “Philosopher’s Stone,” by the agency of which the base metals were to be changed to gold, and the “Elixir of Life,” which was to banish disease and death, were eagerly sought for. Though these were vain imaginings according to modern ideas, nevertheless they were powerful incentives towards experimental work. Many new substances were discovered in this period, and among these were nitric acid (aqua fortis), hydrochloric acid (spirit of salt), and sulphuric acid (oil of vitriol).
Acids were then valued above all other substances. The mediaeval chemist (or alchemist, as he was called) clearly saw that unless a body could be dissolved up there was no hope of changing it. Nitric acid, therefore, which, in conjunction with hydrochloric acid, dissolved even gold itself, was very highly esteemed. Oil of vitriol also was scarcely less important, for it was required for the production of other acids.
So far, taste and solvent power were considered to be the characteristic feature of acids. In the time of Robert Boyle (1627-1691), they were further distinguished from other substances by the change which they produced in the colour of certain vegetable extracts. Tincture of red cabbage was first used, but, as this liquid rapidly deteriorates on keeping, it was soon replaced by a solution of litmus, a colouring matter obtained from Roccella tinctoria and other lichens. It imparts to water a purple colour, which is changed to red by the addition of acids.
Alkalis. Wood ashes were valued in very early times because they were found to be good for removing dirt 3 from the skin. Mixed with vegetable oil or animal fat, they formed a very primitive kind of soap, which was afterwards much improved by using the aqueous extract instead of the ashes themselves, and also by the addition of a little caustic lime.
When plant ashes are treated with water, about 10 per cent. dissolves. If the insoluble matter is then allowed to settle down and the clear liquid evaporated to dryness, a whitish residue is obtained. The soluble matter thus extracted from the ashes of plants which grow in or near the sea is mainly soda; that from land plants, mainly potash. Formerly no distinction was made, and the general term “alkali” was applied to both.
In order to bring the properties of alkalis into contrast with those of acids, we cannot do better than make a few simple experiments with a weak solution of washing soda. Its taste is very different from that of an acid; it is generally described as caustic. If a little is rubbed between the fingers, it feels smooth, almost like very thin oil. It does not dissolve metals or limestone. Its action on vegetable colouring matter is just as striking as that of acids. Tincture of red cabbage becomes green; the purple of litmus is changed to a light blue. This colour change is characteristic of alkalis.
Neutralization. When the colour of litmus solution has been changed to red by the addition of an acid, the original colour can be restored by adding an alkali. The change can be repeated as often as desired by adding acid and alkali alternately. From this we get a distinct impression of antithesis between the two. In popular language, an alkali “kills” an acid; in Chemistry, the same idea is expressed by the term “neutralization.”
Salts. Both “neutralization” and “killing the acid” are modes of expression which describe the phenomenon fairly well. When an acid is neutralized, its characteristic taste, its solvent power, and its action on litmus, are all changed; in fact, the acid as an acid ceases to exist, and so does the alkali. When the neutral solution is evaporated to dryness, a residue is found which on examination proves to be neither the acid nor the alkali, but a compound formed from the two. This substance is called a salt.
To most people, salt is the name for that particular substance which is taken as a condiment with food. Its use in this connection dates from time immemorial. It is distinctly unfortunate that another and very much wider usage of the term has been introduced into Chemistry. When the early chemists recognized that other substances, which they vaguely designated as “saline bodies,” were similar to common salt in composition, they took the name of the individual and applied it to the whole class.
Solution of Metals in Acids. Alkalis are not the only substances which neutralize acids. Speaking in a broad and general sense, we may say that an acid is neutralized when a metal is dissolved in it, because, when the point is reached at which no more metal will dissolve, all the characteristic properties of the acid are destroyed. A salt is formed in this case also.
An example will now be given to illustrate this method of salt formation. Before two pieces of metal can be united by soldering, it is necessary to clean the surfaces of the metal and the soldering iron. The liquid used for this purpose is made by adding scraps of zinc to 5 muriatic acid (hydrochloric acid). The zinc dissolves with effervescence, which is caused by the escape of hydrogen gas. When effervescence ceases and no more zinc will dissolve, the liquid is ready for use. The acid has been “killed” or neutralized by the metal. A salt called zinc chloride has been formed. This salt can be recovered from the liquid by evaporation.
Solution of Oxides in Acids. The substances most used in commerce with the express purpose of destroying acidity are quicklime, washing soda, and powdered chalk.
Since quicklime is a compound of the metal calcium and the gas oxygen, its systematic name is calcium oxide; when it neutralizes an acid, it forms the corresponding calcium salt; for example, if it neutralizes acetic acid, calcium acetate is formed.
An instance of the neutralization of an acid by an oxide of a metal is furnished by one method of preparing blue vitriol (copper sulphate). Copper does not dissolve very quickly in dilute sulphuric acid; hence, to make blue vitriol from scrap copper, the metal is first heated very strongly while freely exposed to air. Copper and oxygen of the air combine to form the brownish black powder, copper oxide, and this dissolves very readily in sulphuric acid, making the salt, copper sulphate.
Solution of Carbonates in Acids. Washing soda and chalk belong to a different class of chemical substances. They are carbonates, that is, they are salts of carbonic acid. At first it may seem a little perplexing to the reader to learn that a salt can neutralize an acid to form a salt. It must be remembered, however, that acids differ from one another in strength, that is, in chemical activity, and that carbonic acid is a weak acid. When a salt of carbonic acid—sodium carbonate 6 or washing soda, for example—is added to a stronger acid such as sulphuric acid, sodium sulphate is formed and carbon dioxide liberated.
As an example of the neutralization of acids by carbonates, we may mention here a practical sugar saving device. Unripe fruit is very sour because it contains certain vegetable acids dissolved in the juice. These acids are not affected by boiling; and, therefore, to make a dish of stewed fruit palatable, it is necessary to add sugar in quantity sufficient to mask the sour taste. If a pinch of bicarbonate of soda is added to neutralize the acid, far less sugar will be necessary for sweetening.
Insoluble Salts. The methods given above apply only to those salts which are soluble in water. Insoluble salts are obtained by mixing two solutions, the one containing a soluble salt of the metal, and the other, a soluble salt of the acid or the acid itself.
The formation of an insoluble salt by the interaction of two soluble substances is well illustrated in the preparation of Burgundy mixture, the most effectual remedy yet proposed for checking the spread of potato disease. This mixture contains copper carbonate, that is, the copper salt of carbonic acid. For its preparation we require copper sulphate and sodium carbonate (washing soda), a soluble carbonate. When these two substances, dissolved in separate portions of water, are mixed, copper carbonate is formed as a pale blue solid which is in such a state of fine subdivision that it remains suspended in the solution of sodium sulphate, the other product of the reaction.
The change is represented diagrammatically below. Each circle represents the atom or a group of atoms named therein. At the moment of mixing, these groups undergo re-arrangement.
Bordeaux mixture, which some gardeners prefer, is a similar preparation containing copper hydroxide instead of copper carbonate. It is made by mixing clear lime water (a soluble hydroxide) with copper sulphate.
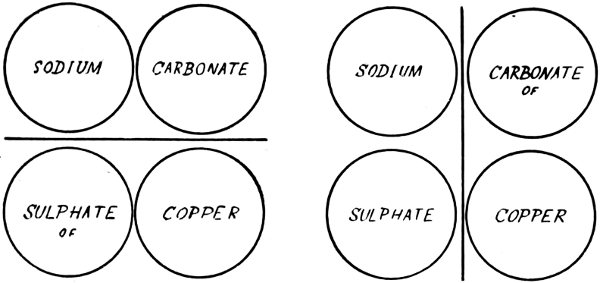
Fig. 1
Elements and Compounds. It is scarcely possible to discuss chemical processes without having from time to time to use terms which are not in everyday use. A few preliminary definitions and explanations of terms which will be frequently used may serve to simplify descriptions, and render it unnecessary to encumber them with purely explanatory matter.
Among the many different kinds of materials known, which in the aggregate amount to several hundreds of thousands, there are about ninety substances which up to the present time have not been broken up into simpler kinds. These primary materials are called “elements,” the remainder being known as “compounds.”
The following is a list of the commonest of these elements, together with the symbols by which they are represented in Chemistry.
| METALS | |
|---|---|
| Aluminium | Al. |
| Antimony (Stibium) | Sb. |
| Barium | Ba. |
| Bismuth | Bi. |
| Cadmium | Cd. |
| Calcium | Ca. |
| Chromium | Cr. |
| Copper (Cuprum) | Cu. |
| Gold (Aurum) | Au. |
| Iron (Ferrum) | Fe. |
| Lead (Plumbum) | Pb. |
| Lithium | Li. |
| Magnesium | Mg. |
| Manganese | Mn. |
| Mercury (Hydrargyrum) | Hg. |
| Nickel | Ni. |
| Platinum | Pt. |
| Potassium (Kalium) | K. |
| Silver (Argentum) | Ag. |
| Sodium (Natrium) | Na. |
| Strontium | Sr. |
| Tin (Stannum) | Sn. |
| Zinc | Zn. |
| NON-METALS | |
|---|---|
| Boron | B. |
| Bromine | Br. |
| Carbon | C. |
| Chlorine | Cl. |
| Fluorine | F. |
| Hydrogen | H. |
| Iodine | I. |
| Nitrogen | N. |
| Oxygen | O. |
| Phosphorus | P. |
| Silicon | Si. |
| Sulphur | S. |
The first step in the building-up process consists of the union of a metallic with a non-metallic element. Such compounds are binary compounds, and are distinguished by the termination -ide added to the name of the non-metallic element; for example, copper and oxygen unite to form copper oxide, sodium and chlorine form sodium chloride, iron and sulphur form iron sulphide or sulphide of iron.
A compound containing more than two elements is distinguished by the termination -ate. Most salts fall within this category; thus we speak of acetate of lead and chlorate of potash, also of sodium sulphate and copper sulphate, the latter form being the more correct.
A difficulty arises when two bodies are composed of the same elements combined in different proportions. Then we have to resort to other distinguishing prefixes or suffixes. For this reason we meet with sulphurous 9 acid and sulphuric acid, the corresponding salts being sulphites and sulphates.
Crystals and Water of Crystallization. When a soluble salt is to be recovered from its solution, the latter is reduced in bulk by evaporation until, either by experience or by trial, it becomes evident that the solid will be formed as the liquid cools. In some cases, when time is not an important factor, evaporation is left to take place naturally. Under either set of conditions, the substance generally separates out in particles which have a definite geometrical form. These are spoken of as crystals.
Crystals often contain a definite percentage of water, called “water of crystallization.” In washing soda, this combined water forms nearly 63 per cent. of the total weight; in blue vitriol, it is approximately 36 per cent. On being heated to a moderate temperature, the water is expelled from the solid; the substance which is left behind is called the anhydrous (that is, the waterless) salt.
Key Industries. The importance of the chemical industries depends mainly on the fact that they constitute the first step in a series of operations by which natural products are adapted to our needs. The materials which are found in earth, air, and water are both varied in kind and abundant in quantity, but in their natural state they are not generally available for immediate use. Moreover, very many substances now deemed indispensable are not found ready formed in Nature.
The end product of the chemical manufacturer is often one of the primary materials of some other industry. Soda ash and Glauber’s salt are essential for making glass; soap could not be produced without caustic alkali; the textile trade would be seriously handicapped if bleaching materials, mordants, and dye-stuffs were not forthcoming. Considered in this light, the preparation of chemicals is spoken of as a “key industry.”
Furthermore, very few of these indispensable substances can be made without using sulphuric acid. This acid is, on that account, just as important to chemical industries as the products of these are to other branches of trade. It may, therefore, be looked upon as a master key of industrial life.
Primary Materials. The composition of sulphuric acid is not difficult to understand. Air is mainly a mixture of oxygen and nitrogen; and when a combustible body burns, it is because chemical action between the material 11 and oxygen is taking place. In this way, sulphur burns to sulphur dioxide. This gas, dissolved in water, forms sulphurous acid, which changes slowly to sulphuric acid by combination with more oxygen. Hence, sulphur, oxygen, and water are the primary materials required for making sulphuric acid.
Sulphur is the familiar yellow solid commonly known as brimstone. It is found native in the earth, and is fairly abundant in certain localities, notably in the neighbourhood of active and extinct volcanoes. Italy, Sicily, Japan, Iceland, and parts of the United States are the principal sulphur-producing countries. Though very plentiful and consequently cheap, only a relatively small quantity of sulphuric acid is made directly from native sulphur, because at the time when this industry was started in England, restrictions were placed on the export of sulphur from Sicily and, consequently, the plant which was then established was adapted to the use of iron pyrites.
Iron pyrites contains about 53 per cent. of sulphur combined with 47 per cent. of iron, and when this is burnt in a good draught, nearly the whole of the sulphur burns to sulphur dioxide, leaving a residue of oxide of iron which can be used for making cast iron of a low grade.
Iron pyrites is often supplemented by the “spent oxide” from the gas works. Crude coal gas contains sulphur compounds which, if not removed, would burn with the gas and form sulphur dioxide. The production of these pungent and suffocating fumes would be a source of great annoyance, and therefore it is necessary to remove the sulphur compounds. To do this, the gas is passed through two purifiers, the first containing slaked lime and the second ferric oxide, both in a slightly moist condition. After being some time in 12 use, the purifying material loses its efficacy; the residue from the lime purifier is sold as “gas lime,” but that from the ferric oxide purifier is exposed to the air and so “revived.” At length, however, it becomes so charged with sulphur that it is of no further use for its original work. It is then passed on to the sulphuric acid maker.
Evolution of the Manufacturing Process. In dealing with the main processes for the manufacture of acids and alkalis, reference will frequently be made to the methods of bygone times. Although as an exact science Chemistry is comparatively modern, as a branch of human knowledge its history goes back to the dawn of intelligence in man. It is agreed that the higher types of living things are more easily understood when those of a simpler and more primitive character have been studied. In like manner, the highly specialized industries of modern times become more intelligible in the light of the efforts of past generations to achieve the same object.
Basil Valentine, who lived in the fifteenth century, states that the liquid which we now call sulphuric acid was in his day obtained by heating a mixture of green vitriol and pebbles. Until quite recent times, sulphuric acid of a special grade was made by precisely the same method, except that the pebbles were dispensed with. In passing, we may remark that the common name “vitriol,” or “oil of vitriol,” is accounted for by this connection with green vitriol. The second method, quoted by Basil Valentine, consisted of the ignition of a mixture of saltpetre and sulphur in the presence of water. This is actually the modern lead chamber process in embryo.
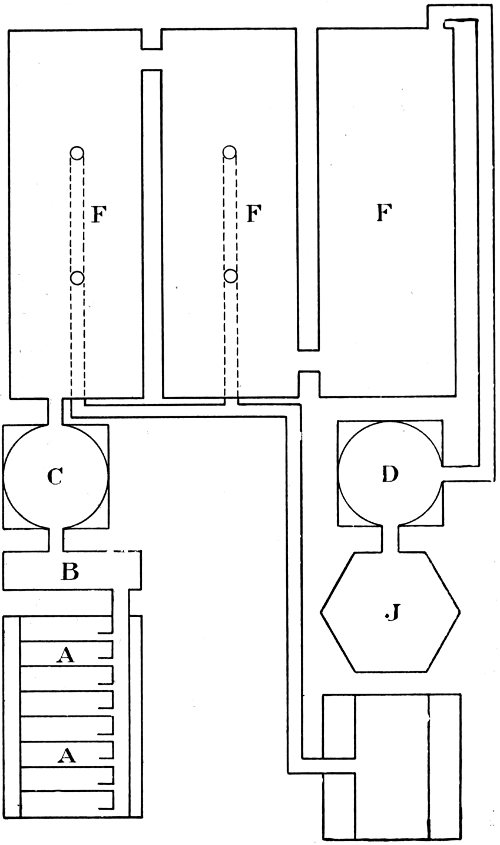
Fig. 2. PLAN OF SULPHURIC ACID WORKS
About the middle of the eighteenth century, “Dr.” Ward took out a patent for the manufacture of sulphuric acid, to be carried on at Richmond in Surrey. He used large glass bell jars of about 40-50 galls. capacity, in which he placed a little water and a flat stone to support a red-hot iron ladle. A mixture of saltpetre and sulphur was thrown into the ladle and the mouth of the vessel quickly closed. After the vigorous chemical action was over, the ladle was re-heated and the process repeated until at last fairly concentrated sulphuric acid was produced.
The large glass vessels used by Ward were costly and easily broken. They were soon replaced by chambers about 6 ft. square, made of sheet lead, but otherwise the process was just the same. The next advance consisted in making the process continuous instead of intermittent. An enormously increased output was thereby rendered possible, and the main features of the modern process gradually developed.
The Lead Chamber Process. We can now consider the actual working of the lead chamber process, aided by the diagrammatic plan of the works shown in Fig. 2. Sulphur dioxide is produced in a row of kilns (A-A) by burning iron pyrites in a carefully regulated current of air. The mixture of gases which leaves the pyrites burners contains sulphur dioxide, excess of oxygen, and a very large quantity of nitrogen. To this is added the vapour of nitric acid, generated from sodium nitrate and concentrated sulphuric acid contained in the “nitre pots,” which are placed at B. The mixture of gases then passes up the Glover tower (C) and through the three chambers in succession, into the first two of which steam is also introduced. Sulphuric acid is actually produced in the chambers, and collects on the floors, from which it is drawn off from time to time. The residual gas from the last chamber is passed up the Gay Lussac tower (D), and after that is discharged into the air by way of the tall chimney (J).

Fig. 3. GENERAL VIEW OF SULPHURIC ACID WORKS
The Oxygen Carrier. We have seen that sulphur dioxide, oxygen, and water are the only substances required to produce sulphuric acid. Why, then, is the nitric acid vapour added to the mixture? As described in a former paragraph, the combining of these gases was represented as being a very simple operation. So indeed it is, for it even takes place spontaneously. 16 Yet, as a commercial process, it would be quite impracticable without the nitric acid vapour, for although the gases combine spontaneously, they do so very slowly, and it is the nitric acid vapour which accelerates the rate of combination.
It is not known with any degree of certainty how the nitric acid acts in bringing about this remarkable change. It has been suggested that reduction to nitrogen peroxide first takes place, and that sulphur dioxide takes oxygen from this body, reducing it still further to nitric oxide, which at once combines with the free oxygen present to form nitrogen peroxide again. So the cycle of changes goes on, the nitrogen peroxide playing the part of oxygen carrier to the sulphur dioxide; and since it is continually regenerated, it remains at the end mixed with the residual gases.
Recovery of the Nitrogen Peroxide. If the gases from the last chamber passed directly into the chimney shaft, there would be a total loss of the oxides of nitrogen, and the consequence of this would be that more than 2 cwt. of nitre would have to be used for the production of 1 ton of sulphuric acid. This would be a serious item in the cost of production, and it is therefore essential that this loss should be prevented.
The recovery of the oxides of nitrogen is effected in the Gay Lussac tower, a structure about 50 ft. in height, built of sheet lead and lined with acid-resisting brick. It is filled with flints, over which a slow stream of cold concentrated sulphuric acid is delivered from a tank at the top. As the gas from the last chamber passes up this tower, it meets the stream of acid coming down. This dissolves and retains the nitrogen peroxide. The acid which collects at the bottom of the tower is known as nitrated vitriol.
The next step is to bring the recovered nitrogen 17 peroxide again into circulation. The nitrated vitriol is raised by compressed air to the top of the Glover tower, and as it trickles down over the flints in this tower it is diluted with water, while at the same time it meets the hot gases coming from the pyrites burners. Under these conditions, the nitrogen peroxide is liberated and carried along by the current of gas into the first lead chamber. The stream of cold acid coming down the Glover tower also serves to cool the hot gases before they enter the first chamber.
In order to complete the description of the works, it is necessary to add a note on the lead chambers themselves. The sheet lead used in their construction is of a very substantial character; it weighs about 7 lb. per square foot. The separate strips are joined together by autogenous soldering, that is, by fusing the edges together. In this way the presence of another metal is avoided; otherwise this would form a voltaic couple with the lead, and rapid corrosion would take place.
The size of the chambers has varied a great deal. In the early years of the nineteenth century, the capacity of a single chamber was probably not more than 1,000 cu. ft.; at the present time, 38,000 cu. ft. is an average size, and there may be three or five of these chambers. The necessity for this large amount of cubic space is easily accounted for. The reaction materials are all gases, and a gas occupies more than one thousand times as much space as an equal weight of a solid or liquid. Moreover, oxygen constitutes only about one-fifth of the total volume of air used in burning the pyrites; the other four-fifths is mainly nitrogen, which, though it does not enter into the reaction at all, has to pass through the chambers.
Modern Improvements. Among the modern innovations in the lead chamber process, the following are 18 worthy of note. “Atomized water,” that is, water under high pressure delivered from a fine jet against a metal plate, has certain advantages over steam. In order to bring about a more rapid mixing of the gases in the chamber, it is proposed to make these circular instead of rectangular, and to deliver the gases tangentially to the sides. Another suggestion is to replace the lead chambers by towers containing perforated stoneware plates set horizontally. By this arrangement, since the holes are not placed opposite one another, the gases passing up the tower must take a zig-zag course. This makes for more efficient mixing.
Sulphur Trioxide. When elements are combined in different proportions by weight, they produce different compounds. Thus, in the case of sulphur and oxygen, there are two well-known compounds, namely, sulphur dioxide and sulphur trioxide. In the former, a given weight of oxygen is combined with an equal weight of sulphur; in the latter, this same weight of sulphur is combined with 50 per cent. more oxygen. On this account, sulphur trioxide is spoken of as the higher oxide.
We can now state in general terms another method by which sulphuric acid can be built up from its elements. Sulphur, as we have seen, burns in oxygen, forming sulphur dioxide. This substance can then be made to unite with more oxygen to give sulphur trioxide, which, with water, yields sulphuric acid. There are three steps in this synthesis. The first, namely, sulphur to sulphur dioxide, has already been considered; the last, sulphur trioxide to sulphuric acid, only requires that sulphur trioxide and water shall be 19 brought together: we can, therefore, confine our attention to the intermediate step, namely, the conversion of sulphur dioxide into trioxide.
This operation, when carried out in a chemical laboratory, is a very simple one. Fig. 4 shows the necessary apparatus. Sulphur dioxide from a siphon of the liquefied gas and air from a gasholder are passed into the Woulff’s bottle A, containing concentrated sulphuric acid; this removes moisture from the gases. The drying process is completed in the tower B, which contains pumice stone soaked in sulphuric acid. The mixed gases then pass through the tube C, containing platinized asbestos heated to about 400° C.: the sulphur trioxide collects in the cooled receiver D.
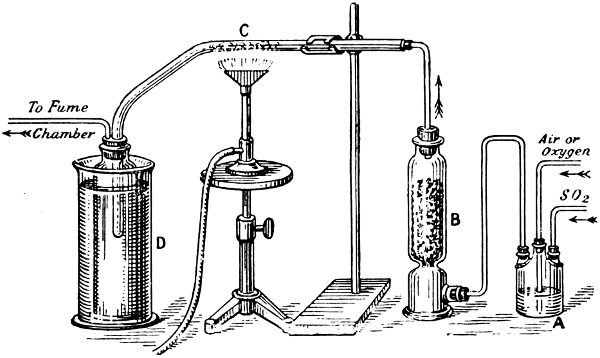
Fig. 4. SULPHUR TRIOXIDE—THE CONTACT PROCESS
Platinized asbestos is made by soaking long-fibred asbestos in a solution of platinum chloride. The material is then dried and subjected to a gentle heat. 20 In this way, metallic platinum in an exceedingly fine state of subdivision is deposited on the asbestos fibre, which merely serves as a convenient support.
Catalytic or Contact Action. The influence of the finely divided platinum is a very important factor in the reaction. It cannot, however, be said to cause the union of sulphur dioxide with oxygen, for the gases combine to a very slight extent when it is not present. What the platinum actually does is to influence the rate of formation to such a degree that, under favourable conditions, practically the whole of the sulphur dioxide is changed to sulphur trioxide instead of an exceedingly small fraction of it.
The most interesting, and at the same time the most perplexing, feature of the reaction is that the platinum itself does not appear to undergo any change. It is not diminished in quantity, for only a very small amount is necessary for the conversion of a very large amount of the mixed gases. Its activity lasts for a very long time, and even when it does become inactive, it can be shown that this is due to some external cause, such as the presence of dust and certain impurities in the gases.
Many other similar cases are known in which the presence of a small quantity of a third substance greatly influences the course of a chemical reaction without appearing in any other way to be necessary to the reaction. These substances, which are often metals in a very fine state of subdivision, are called catalytic or contact agents.
The Contact Process for making sulphuric acid is nothing more nor less than the simple laboratory operation which we have described above, carried out on a larger scale.
The sulphur dioxide is produced as in the lead chamber process by roasting iron pyrites in a current 21 of air. This gas, together with the excess of air, is passed into the contact furnace, which consists of four tubes, each containing platinized asbestos, supported on perforated plates. The union of the two gases is said to be almost complete: an efficiency of 98 per cent. of the theoretical value is claimed for this process. The sulphur trioxide, or “sulphuric anhydride”[1] is either condensed in tin-lined drums or absorbed in ordinary concentrated sulphuric acid.
The proposal to manufacture sulphuric acid by this method was first made in 1831 by Peregrine Phillips, of Bristol. The early attempts were not successful, and it was not until about forty-four years later that the difficulties arising in the working of the contact process were overcome sufficiently to enable the sulphuric acid produced in this way to be sold at the same price as that made by the lead chamber process. Since 1890, the total quantity of acid made by the contact method has increased very rapidly, so that it now furnishes about one-half of the world’s supply, and seems likely in time to displace the lead chamber process altogether.
The history of the rise of the contact process is interesting because it illustrates in a striking manner the very great difference that there is between a successful laboratory process and a successful manufacturing process, though seemingly identical.
The first and possibly the most serious difficulty encountered in the working of the contact process was the frequent interruption caused by the loss of activity of the contact substance. Iron pyrites always contains arsenic which volatilizes on heating, and this quickly caused the platinum to lose its activity, or, as it was sometimes rather fancifully expressed, “poisoned the 22 catalyst.” Dust also is inevitable, and this, carried forward mechanically with the stream of gas, settled on the contact substance and caused the action to cease.
To get over this difficulty it is necessary to purify the gases. They are first passed slowly through channels in which the coarser particles of dust settle down. Steam is injected into the mixture to wash out the finer particles of solid, and also to get rid of arsenic, and then the gases are passed through scrubbers. Before being admitted to the contact furnace, the moist gas is submitted to an optical test. It is passed through a tube, the ends of which are transparent; a bright light is placed at one end and viewed from the other through a column of gas of considerable length. If the purification process is working satisfactorily, there is a complete absence of fog. The gases are then dried by passing through concentrated sulphuric acid and admitted to the contact tubes.
In all operations carried out on a large scale, the regulation of temperature is a matter of some difficulty. In the case which we are considering, the most suitable temperature range is a rather narrow one, and the difficulty of keeping within the limits is very much increased by the large amount of heat given out when the sulphur dioxide and oxygen combine. The result of the failure to maintain the temperature at a fairly constant level was that the process worked in a very irregular manner, for as soon as it was working really well and sulphur trioxide was being formed rapidly, the heat given out by the reaction itself was also great, and consequently, the higher temperature limit was exceeded.
The method of controlling the temperature in the contact process is worth noting, because it is really ingenious. The tubes containing the platinized asbestos are surrounded by wider concentric tubes. The gases 23 which are about to enter the contact furnace pass through the annular space between the two tubes, and are thereby heated to the required temperature, while at the same time they serve to cool the inner tubes. The most satisfactory temperature is about 400° C. The tubes are first warmed to 300° C. to start the reaction, and thereafter the heat evolved by the reaction itself is sufficient to keep it going.
The absorption of the sulphur trioxide also caused some difficulty at first. This substance reacts most violently with water, dissolving with a hissing sound like that produced when a red-hot poker is plunged into water. At the same time great heat is developed, and consequently, much of the sulphur trioxide is vaporized, and in that way lost. This difficulty was got over by using 98 per cent. sulphuric acid for the absorption, the acid being kept at this strength by the simultaneous addition of water.
The contact process has some very distinct advantages over the older lead chamber process. The plant covers a much smaller area than the bulky lead chambers. Although the preliminary purification of the gases is somewhat tedious and costly, this is in great measure compensated by the purity of the acid produced. No separate plant is required for concentration and purification, as in the older process. Finally, sulphuric acid of any concentration can be produced at will, including the fuming acid, which is required as a solvent for indigo, and in the manufacture of artificial indigo and other organic chemicals.
The lead chamber process produces what is called chamber sulphuric acid very cheaply. Although this is only a 60-70 per cent. solution and very impure, nevertheless, it is quite good enough for the heavy chemical trade, particularly for the first stage of the 24 Leblanc soda process, and for making superphosphate. These two industries alone consume many thousands of tons of this sulphuric acid every year. Probably for some years to come the two processes will continue to exist side by side, but it may be doubted whether new works will now be installed to make sulphuric acid by the lead chamber process.
Properties of Sulphuric Acid. The pure non-fuming acid is a colourless oily liquid whose density is 1·84. It mixes with water in all proportions, yielding dilute sulphuric acid, and it also dissolves sulphur trioxide, yielding the fuming acid.
The mixing of sulphuric acid and water is accompanied by an evolution of heat and by contraction in volume. It is an operation which must be carried out with great care, the acid being always poured into the water, otherwise the water floats on the heavier acid, and so much heat is developed at the surface of separation that some of the water will be suddenly converted into steam, and this, escaping from the liquid with explosive violence, may cause the contents of the vessel to be scattered about.
Strong sulphuric acid chars most organic substances. From substances such as wood, sugar, paper, starch, it withdraws the elements of water, liberating carbon. Since it acts in the same way upon human flesh, it is clear that the concentrated acid must be handled with very great care, for it causes most painful burns. For this reason, vitriol throwing has always been regarded as a most serious and dastardly offence. A simple first-aid remedy for burns produced by sulphuric acid is the liberal application of an emulsion of linseed oil and lime water. The lime, being an alkali, neutralizes the acid, and the oil excludes air from the wound.
The readiness with which sulphuric acid combines 25 with water is often made use of both in the laboratory and in industrial Chemistry for the purpose of drying gases. One illustration of this use has already been given in describing the contact process. Another instance which may be fairly familiar occurs in the case of liquefying air, where the gas must be thoroughly dried before being passed into the refrigerating apparatus, otherwise this would soon become blocked with ice.
The position which sulphuric acid occupies in Chemistry is due mainly to three outstanding features. In the first place, it is a strong mineral acid and displaces all other acids from their salts. Secondly, it has a high boiling point (338° C.), and consequently, the displaced acid with the lower boiling point can be distilled from the mixture. Lastly, sulphuric acid can be made very cheaply from materials which are very abundant in Nature, and, therefore, it meets all the requirements of an acid which is to be used for general purposes.
All the common metals, except gold and platinum, dissolve either in concentrated or in dilute sulphuric acid, forming sulphates. These salts are highly important and interesting substances. They are all soluble in water, with the exception of the sulphates of calcium, strontium, barium, and lead.
Ferrous Sulphate, also called green vitriol and copperas, is obtained by dissolving iron in dilute sulphuric acid. The solution is green, and when it is evaporated, the crystals which separate out look like bits of green glass. It was because of this that the substance was first called green vitriol (vitrum = glass). It is used very largely in dyeing as a mordant. Writing ink and Prussian blue are also made from it.
The Alums are double sulphates. They are made by crystallizing solutions of potassium, sodium, or ammonium sulphate together with solutions of iron (ferric), chromium, or aluminium sulphates. In this way, we may have potassium aluminium alum, or iron ammonium alum, and so on, but whichever combination of elements is present, the salt which is formed always crystallizes in octahedra. The chief use of the alums, as also of aluminium sulphate, is as mordants in dyeing.
Since a great many metallic salts, particularly acetates and sulphates, are used in the dye industry as mordants, it may be well to explain here very briefly what a mordant is.
It must be remembered that almost all the dyes are solids which dissolve in water, yielding intensely coloured solutions. Hence, in most cases, if a fabric is merely dipped in the dye and then dried, the colouring is not permanent, but can be washed out with water. In order to fix the colouring matter, the material is first dipped in the mordant, usually a bath of some metallic salt, and then, generally after exposure to air or after steaming, into the dye bath, with the result that the colour becomes fixed. The first part of the process is called “mordanting” the material. The mordant either adheres to or combines with the fibres, and the dye forms with the mordant a coloured compound called a “lake,” which resists the action of water. The colour is then said to be “fast,” that is, firmly fixed.
For printing on calico, the mordant is thickened with gum arabic or other glutinous substance. The design is then stamped on the material with the thickened mordant liquor. The subsequent treatment consists of dipping the material in the dye and afterwards in water, when the colour comes away from those parts which have not received the impress of the mordant.
Sodium Sulphate, or Glauber’s salt, is made from common salt by the action of concentrated sulphuric acid. It is one of the raw materials used in making glass.
Ammonium Sulphate. (See p. 99.)
Calcium Sulphate, or gypsum, occurs in large quantities in Nature. The salt contains 20·9 per cent. of combined water, and when carefully heated to 120° C, it loses about two-thirds of this water, yielding a white powder known as plaster of Paris. This substance, when made into a paste with water, gradually sets to a hard mass, because the partially dehydrated gypsum re-combines with the water.
Lead Sulphate, the chief impurity of commercial oil of vitriol, is a white powder which is very often used for making white paint in place of lead carbonate (white lead). The sulphate has the advantage over the carbonate in not being so readily discoloured; its disadvantage is that it lacks “body.”
Copper Sulphate, or blue vitriol, is frequently found in the drainage of copper mines, where it is formed by the oxidation of copper pyrites. It is made on a large scale by roasting sulphide ores of copper in a current of air. Oxygen combines with copper sulphide, forming copper sulphate, which is extracted with water and crystallized. It forms large blue crystals containing 36 per cent. of water. This salt is put to many different uses. Very large quantities are used for dyeing and calico printing; some of the green pigments, such as Schweinfurt green, are made from it.
Nitric acid, the aqua fortis of the alchemists, must be placed next to sulphuric acid in the scale of relative importance, because of the variety of its uses. It is indispensable for making explosives, and is used for the preparation of drugs and fine chemicals, including the coal-tar dyes. The acid also dissolves many metals, forming nitrates, which are put to several uses. Silver nitrate is the basis of marking ink, and it is also the substance from which the light-sensitive silver compounds required for the photographic industry are made. The important pigments, chrome yellow and chrome red, are prepared from lead nitrate. The solvent action of nitric acid on copper is made use of in etching designs on copper plates. Over and above all this, it must be mentioned that an adequate supply of “nitrate” is required for artificial manure. Thus it can be said that with the uses of this acid and its salts are associated our supply of daily bread, our freedom from foreign oppression, and many of the refinements and conveniences of life.
We shall begin the study of nitric acid by taking stock, as it were, of the natural sources of supply. The free acid is not found in Nature except for very small traces in the air after thunderstorms. We have, therefore, to rely entirely on that which can be obtained artificially. Until quite recently, it could be said that there was only one method of making the acid, namely, by the 29 distillation of a mixture of potassium or sodium nitrates and concentrated sulphuric acid. Now, however, nitric acid is being made from the air, though as yet only in small quantity, notwithstanding the great development of this method owing to war requirements; hence, we are still mainly dependent on the naturally occurring nitrates just mentioned.
Potassium Nitrate (nitre, saltpetre, sal prunella) is found in the soil of hot countries, especially in the neighbourhood of towns and villages where the sanitary arrangements are primitive. In very favourable circumstances, it may even appear as a whitish, mealy efflorescence on the surface of the ground. To obtain the salt, it is only necessary to agitate the surface soil with water and, after the insoluble matter has settled down, to evaporate the clear solution.
Potassium nitrate is required for making gunpowder, which, until quite recent times, was the only explosive used in warfare. Continental countries that could not afford to rely entirely on sea-borne nitre had to make their own. The refuse of the farmyard, mixed with lime and ashes, was made up into a heap of loose texture, which was periodically moistened with the drainage from the stables. In the course of years, saltpetre and calcium nitrate were formed in the surface layers, from which they were extracted from time to time. The farmer was then allowed to pay part of his taxes in nitrates.
Sodium Nitrate, also called caliche, Chili-saltpetre, or Chili-nitrate, comes mainly from South America. The beds extend for a distance of about 220 miles in Chili, Peru, and Bolivia, between the Andes mountains and the sea. The deposit is about 5 ft. thick, and its average breadth 5 miles. The crude material is treated with water in steam-heated wooden vats. The clear 30 solution is evaporated, and the residue obtained is washed with the mother liquor and dried. This product may contain as much as 98 per cent. of the nitrate.
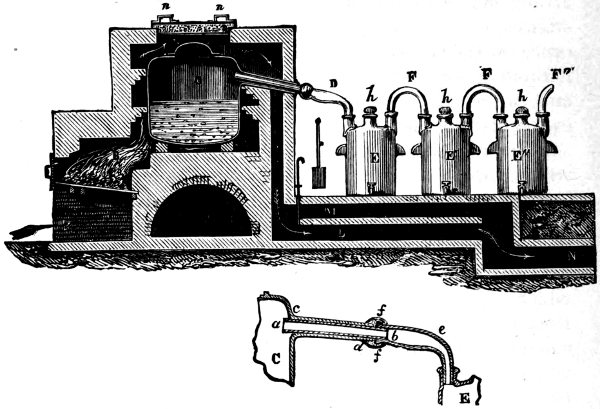
Fig. 5. PREPARATION OF NITRIC ACID
Nitric Acid. Chili-nitrate is always used for making nitric acid. It is the more abundant of the two naturally occurring nitrates, and therefore cheaper; moreover, weight for weight, it yields more nitric acid than the corresponding potassium compound. A mixture of sodium nitrate and sulphuric acid is heated in a large cast-iron retort (C, Fig. 5). The retort is entirely surrounded by flame and hot gases to prevent the condensation of the acid on the upper parts. If this precaution were not taken, the acid would dissolve the iron and the life of the retort would not be long; moreover, the product would contain ferric nitrate as an impurity. 31 The vapour of the acid is led away by the tube D into a series of two-necked earthenware receivers called bonbonnes (E), and there condenses to a liquid. The lower figure shows how the leading tube of the retort is protected from corrosion by the clay tube a, b; and how it is connected to the first receiver by the glass tube e, which is luted on at f. The percentage strength of the acid which distils over depends upon that of the sulphuric acid used and on the purity of the sodium nitrate.
Pure nitric acid is a colourless liquid 1·559 times as heavy as water, volume for volume. It fumes strongly in air, and is a very corrosive liquid. The pure acid of commerce is obtained by distillation of a less concentrated acid. It is 68 per cent. pure. It is rendered free from dissolved oxides of nitrogen by blowing air through it. When kept exposed to light, the colour changes at first to yellow and then to brown, because light causes a certain amount of decomposition.
Red fuming nitric acid owes its colour to the great quantity of oxides of nitrogen dissolved in it. It is made by distilling sodium nitrate that has been thoroughly dried with the strongest sulphuric acid; the distillation is carried out at a high temperature, with the express purpose of decomposing some of the nitric acid to furnish the oxides of nitrogen. Sometimes a little powdered starch is also added to facilitate the formation of these oxides. This variety of nitric acid is particularly active and is used in many operations, especially in making dyes, explosives, and other organic chemicals.
Nitric acid has all the general properties of an acid, that is, it has a sour taste even in very dilute solution, it changes the colour of litmus to red, and dissolves carbonates and many metals.
When the vapour of nitric acid is passed through a red-hot tube, and also when a nitrate is strongly heated, oxygen gas is given off. Analysis shows that the oxygen combined in pure nitric acid amounts to 76 per cent. of its weight, while that in sodium and potassium nitrates is 56 and 50 per cent. respectively. Nitric acid and the nitrates are, therefore, highly oxygenated compounds; moreover, under favourable circumstances, they are rather easily broken up.
Pure nitric acid will set fire to warm, dry sawdust, and a piece of charcoal or sulphur thrown on the surface of molten nitre takes fire spontaneously and is quickly consumed, giving out a very vivid light. The explanation of this is that the supply of oxygen is abundant; it is also readily available and concentrated in a small space. We can vary the experiment. When a mixture of 75 parts by weight of finely-powdered saltpetre, with 15 of charcoal dust and 10 of ground sulphur, is ignited, it burns very vigorously, and is soon consumed. This mixture is, indeed, home-made gunpowder.
Explosives. Gunpowder was discovered in very early times by the Chinese, but for many years the secret of its composition did not get outside the Great Wall. In the fifth century A.D., it was apparently re-discovered at Constantinople, and that city was for a long time defended by the use of what is known in history as Greek Fire, an incendiary mixture very similar to, if not actually the same as, gunpowder. But again the secret of its composition was jealously guarded, and it was not until the thirteenth century that it was discovered, apparently for the third time, and introduced to Western Europe by Roger Bacon. It was used in siege cannon early in the fourteenth century and in field guns at Crécy; but it was apparently not employed for blasting until about 1627, although in 1605, Guy 33 Fawkes and his fellow-conspirators were able to obtain it in large quantity.
From the battle of Crécy in 1346 to the beginning of the South African campaign in 1889, gunpowder was the only explosive used in warfare. “Villainous saltpetre” has therefore played a very important part in shaping the course of events in the world’s history. At the present day, gunpowder has become “old-fashioned.” In warfare, it has been superseded by “smokeless” powders of much greater power; while for mining operations, explosives with a much greater shattering effect have long since taken its place.
The composition of gunpowder may vary, but on the average it contains 75 parts by weight of saltpetre to 15 of charcoal and 10 of sulphur. It is, therefore, a mixture of two combustible substances, with a large quantity of a third very rich in oxygen. The separate constituents are very finely ground and afterwards thoroughly incorporated. When the mixture is ignited, charcoal and sulphur burn very fiercely in the oxygen supplied by the saltpetre.
The secret of the action of gunpowder lies in the extraordinary rapidity with which combustion, started at one point, is propagated through the whole mass. Moreover, the products of combustion are mainly gases, and these occupy several thousand times the volume of the solid from which they are produced. In a confined space, a gas may exert enormous pressure when its normal tendency to expand is resisted.
Propellants. Although combustion is propagated through a quantity of gunpowder with very great rapidity, it is not done instantaneously. The time required is about one-hundredth of a second under ordinary conditions, and this interval, short though it is, is very important. When the object is to throw 34 a projectile, the inertia of the latter has to be overcome, that is, a certain amount of force has to be applied before the heavy body begins to move. In order that the strain on the breech of the gun may be as small as possible, the pressure must be gradually developed and must reach its maximum just as the projectile begins to move.
The time factor in the explosion constitutes the difference between what we now call “propellants” and “high explosive.” Propellants are explosives which develop pressure gradually, and are therefore used to launch the projectile; high explosive develops pressure instantaneously, and is therefore used as the bursting charge inside the shell, bomb, or torpedo, and also in blasting operations.
Cordite, or smokeless powder, is the propellant now most used. It is made by macerating guncotton and nitroglycerine with their common solvent acetone. A pulp is thus made to which 5 per cent. of vaseline is added. The mixture is then forced through a die, and in this way it is formed into threads or rods, which harden as the acetone evaporates. Cordite produces no smoke, because all the products of its combustion are invisible gases.
High Explosive. Nitroglycerine and Guncotton are both explosives of the instantaneous kind. The former is made by forcing glycerine, under pressure in a very fine stream, into a mixture of fuming nitric and concentrated sulphuric acids; the latter by soaking cotton-wool in a similar mixture. Both products are washed with water until quite free from acid, and subsequently dried.
Nitroglycerine is a colourless oil with a burning taste. The oil itself is very dangerous to handle, for it is liable to explode spontaneously even when the utmost care 35 has been taken in its preparation. A mere spot on a filter paper explodes with a deafening report when gently hammered on an anvil; and one drop, when heated on a stout iron plate, blows a hole through the plate. No use could be made of this substance for many years after its discovery because it was so liable to explode during transportation; now, however, it is made safer by mixing with absorbent infusorial earth or kieselguhr. This mixture is known as dynamite. Blasting gelatine, like cordite, is a mixture of nitroglycerine and guncotton.
Trinitrotoluene (T.N.T.) is made from toluene and nitric acid, and is a type of the modern high explosive. It is a yellow crystalline substance which melts at 79°-81·5° C., and is poured into the shell in a molten condition. It is a remarkably stable substance, which burns quickly when heated to 180° C.; it cannot be exploded even by hammering. Explosion is only brought about by that of a subsidiary substance called the detonator. The percentage composition of T.N.T. is as follows—
| Carbon | 33·5 |
| Hydrogen | 2·3 |
| Nitrogen | 19·5 |
| Oxygen | 44·7 |
| 100·0 |
The oxygen present is only just sufficient to burn the whole of the carbon to carbon monoxide; but since carbon dioxide is also formed, which requires twice as much oxygen for the same weight of carbon, and since the hydrogen and nitrogen may also be oxidized, the combustion of the carbon is not complete; and therefore the explosion of T.N.T. is accompanied by a dense 36 black smoke, consisting of finely divided particles of carbon.
The explosive known as ammonal is a mixture of T.N.T., aluminium powder, and ammonium nitrate; the function of the latter substance is to supply more oxygen to render the combustion of the carbon of T.N.T. complete.
Nitrates and the Food Supply. Chemical analysis shows that compounds of nitrogen enter largely into the composition of the living tissues of all plants and animals; hence, either nitrogen itself or some of its compounds must be assimilated by all living organisms to provide for growth and development, and to repair wastage. Air, since it contains approximately four-fifths of its volume of free nitrogen, is the most obvious source of supply. At every breath, a mixture of oxygen and nitrogen is inhaled by animals, but only part of the oxygen is used. Practically the whole of the nitrogen is returned to the atmosphere unchanged; it serves only to dilute the oxygen. From this it is clear that animals do not build up their nitrogenous constituents from elementary nitrogen.
With plants it is very much the same, for, although they obtain their principal food, namely, carbon, from the carbon dioxide which is present in air, it is only in a few exceptional cases that free nitrogen is assimilated. The exceptions will be considered first, because it was through these that we first began to learn something definite about the great importance of nitrogen in agriculture.
Virgil, who was born in 70 B.C., wrote a poem in praise of agriculture. Almost in the opening lines he deals with the treatment of corn land. He advises that, in alternate years, this should either be left fallow or sown with pulse, vetch, or lupin; but not with flax or 37 oats, because they exhaust the land. From this we learn that rotation of crops was one of the established principles of good husbandry even at the beginning of the Christian era.
It was not until the later years of the nineteenth century that any explanation as to why rotation of crops is beneficial was put forward. Let us first state the facts more precisely. Peas, beans, vetches, clover, and other members of the natural order called Leguminosae, which includes about 7,000 species, produce fruits rich in complex nitrogen compounds without being dependent in any way upon nitrogen compounds in the soil. Moreover, they do not exhaust the land as far as these compounds are concerned; hence wheat and other grain can be grown on the same land the following year.
It is now known that leguminous plants assimilate atmospheric nitrogen with the help of certain bacteria. If anyone will dig up a lupin root, he will observe[2] conspicuous wrinkled swellings or nodules at various points on the roots. These, when examined with a high-power microscope, are found to contain colonies of bacteria. It is these minute vegetable organisms which assimilate nitrogen and pass on nitrogen compounds to the larger plant. Other plants cannot assimilate what we might call raw nitrogen; they require soluble nitrates. These they build up into complex organic nitrogen compounds suitable for the feeding of animals which can assimilate neither free nitrogen nor nitrates.
The Nitrogen Cycle. The supply of nitrates in the soil needs continually to be renewed by the addition of decaying vegetable matter, stable or farmyard manure, or Chili saltpetre. The natural manures contain organic nitrogen compounds which were built up during the life of some animal or plant. They are not immediately 38 available as food for other plants, because they are, as it were, the end products of life, and are not soluble in water. But Nature provides for this. The manures decay, forming humus, and ultimately ammonia, one of the simplest of inorganic nitrogen compounds. Ammonia is then transformed to nitrites by certain bacteria present in the soil, while other bacteria change nitrites into nitrates. Both of these organisms are quite distinct from the root nodule bacteria of the Leguminosae.
The nitrates pass into the plant in solution, and then begins again that wonderful cycle of changes which we have described. This is perhaps made clearer by the following diagram.
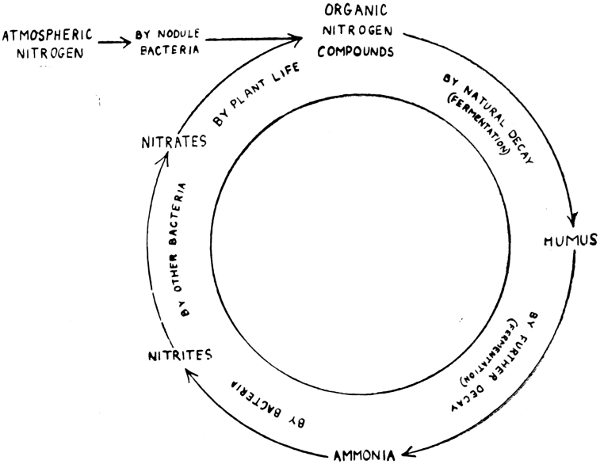
Fig. 6. THE NITROGEN CYCLE
It now remains to show why artificial manures also are necessary. Let us consider what happens to a piece of ground which is left uncultivated. Although nothing is taken from it in the way of a crop, yet it very quickly 39 deteriorates, and the soil becomes infertile through the loss of nitrogen compounds. This is explained by the fact that nitrates are soluble in water, and so they get washed away from the top soil. In addition to this, the nitrogen which is returned to the land forms quite an insignificant fraction of that which is taken from it, for we waste a great deal of organic nitrogen. The difference on both these accounts has, therefore, to be made up by the addition of artificial manures containing soluble nitrates.
The natural supply of nitrate is very limited. According to a report of the Chilian Government published in 1909, the nitre beds of that country were expected to last for less than a century at the current rate of consumption. Wheat, above all things, will not grow to perfection on soil which is deficient in nitrate. In 1908, Sir William Crookes called attention to the difficulty which might be experienced in the near future in supplying the people of the world with bread. Statistics showed that wheat was grown on 159,000,000 acres out of a possible 260,000,000. The average yield is 12·7 bushels per acre. By 1931, it is calculated that the population of the world will be 1,746,000,000; and to supply these with bread, wheat would have to be grown on 264,000,000 acres, that is, 4,000,000 acres beyond the total available wheat land.
The remedy which Sir William Crookes suggested in order to avoid famine was to raise the average yield from 12·7 to 20 bushels per acre by the application of an additional 12,000,000 tons of Chili saltpetre per annum. In view of the possible exhaustion of the supply of this substance, this would only mean a postponement of the evil day. The position, however, is now modified to a great extent because undeveloped deposits of sodium nitrate are known to exist in Upper 40 Egypt, and the making of nitric acid from the air, which in 1908 was put forward as a suggestion, is now an accomplished fact.
Nitric Acid from Air. The supply of nitrogen in the air is truly inexhaustible; it amounts to about 7 tons for every square yard of the earth’s surface, which is about 200,000,000 square miles. It is quite evident that anything man may do in the way of taking nitrogen from the air will make no perceptible difference to its composition.
Every time a flash of lightning passes between a cloud and the earth, oxygen and nitrogen combine in the path of the spark, producing oxides of nitrogen. These dissolve in water, and are washed into the earth as a very dilute solution of nitric acid. As long ago as 1785, H. Cavendish imitated this natural phenomenon. A reference to the diagram (Fig. 7) will show how nitric acid can be made from the air on a small scale. The globe contains air under slightly increased pressure. The platinum wires or carbon rods are connected with the terminals of an induction coil, which in its turn is connected to accumulators supplying the current required.
When the coil is put into action, a spark passes across the gap between the ends of the carbon rods. With a larger coil and a more powerful battery, there is an arching flame which can be blown out and re-lighted. This is actually nitrogen burning in oxygen. The result in either case is the same; the air in the globe sooner or later acquires a reddish-brown colour due to oxides of nitrogen, which, when shaken with water, form a very dilute solution of nitric acid.
The same process is now carried out on a large scale. Air is driven by fans through a very powerful electric arc, whereby 1·5 to 2 per cent. is converted into nitric oxide. This combines spontaneously with more oxygen to form nitrogen peroxide, which, when dissolved in water, gives a very dilute solution of nitrous and nitric acids.
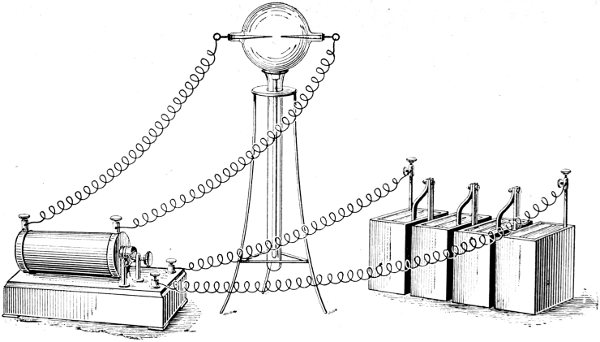
Fig. 7. NITRIC ACID FROM AIR
The absorption of the oxides of nitrogen is carried out systematically. The mixed gases, after passing through the arc, are passed through a series of towers filled with acid-resisting material over which a stream of water is flowing. The solution of nitric acid so obtained is very dilute, but by using the liquid over and over again, a moderately strong solution is ultimately produced. This is collected in granite tanks and neutralized with lime, forming calcium nitrate or Norwegian saltpetre, as it is now called.
This is a new industry and a rapidly-growing one; in the course of five years (1905-1909) the annual output of Norwegian or “air” saltpetre increased from 115 to 9,422 tons. Mountainous countries like Norway and Switzerland are perhaps in a specially favoured position with respect to this industry. Rapid streams and waterfalls, in conjunction with turbines, are used for driving the dynamos, and in this way electricity is produced at very low cost. It is interesting, however, to note that a plant for the manufacture of nitric acid from air has now been established in Manchester.
A group of acids, namely, hydrochloric, hydrofluoric, hydrobromic, hydriodic, must now be considered together with their corresponding salts. In appearance and in other physical properties they resemble one another very closely; they are, therefore, called by the general name “halogen acids.” This name is derived from the Greek word meaning “sea-salt,” which is a mixture of the salts of these acids, and from which the acids themselves can be obtained by treatment with oil of vitriol.
Hydrochloric Acid. When concentrated sulphuric acid is added to common salt, a gas is liberated which has a very pungent acid smell and taste. This is a compound of the elements hydrogen and chlorine, and therefore called hydrogen chloride. It is extremely soluble in water; a given volume of water dissolves as much as 500 times its own volume of the gas. The solution produced in this way is now called hydrochloric acid, but formerly it was known as spirits of salt, or muriatic acid.
Hydrochloric acid has all the general properties of acids. It dissolves many metals, such as zinc, iron, aluminium, and magnesium; hydrogen gas is given off, and the chloride of the metal is formed. It also dissolves limestone, marble, and all forms of calcium carbonate; carbon dioxide gas is liberated, and a solution of calcium chloride remains.
The hydrochloric acid of commerce is obtained as a 44 by-product in the manufacture of washing soda from common salt by the method proposed by Nicholas Leblanc towards the end of the eighteenth century. In the first stage of this process, salt is mixed with sulphuric acid; this causes the liberation of hydrogen chloride gas, which, when dissolved in water, produces hydrochloric acid.
The past history of this branch of chemical industry is interesting. Until about 1870, there was no very great demand for hydrochloric acid, and in the early days of the working of the Leblanc process the soda manufacturer took no pains to recover more than he could actually sell. Consequently, a large quantity of hydrogen chloride gas was allowed to escape into the air, with results which can well be imagined. For miles around, great damage was frequently sustained by the growing crops; when it rained in the neighbourhood of the works, the gas was washed out of the air and, speaking quite literally, it rained dilute hydrochloric acid, which rapidly corroded all stone and metal work. It is not, therefore, surprising to learn that alkali makers were frequently involved in litigation, and chemical works were regarded as a great nuisance.
By the Alkali Act of 1863, chemical manufacturers were compelled to prevent the escape of more than 5 per cent. of hydrochloric acid gas; and by a subsequent Act, this limit was lowered to 0·2 grain per cubic foot. The provisions of the Acts were not difficult to carry out, because hydrogen chloride is extremely soluble in water.
The gases coming from the pans in which the salt was decomposed were led into towers (see Fig. 8) built of bricks or Yorkshire flags soaked in tar. These towers were filled up with coke or other acid-resisting material, which was kept moist by water flowing from the tank F. 45 In this way, hydrogen chloride gas was removed and hydrochloric acid collected in tanks (not shown in the figure) at the bottom of the towers. Even then, there was no market for the greater part of the recovered acid, consequently much of it found its way into drains and streams, and so carried on its work of destruction in a less obtrusive way.
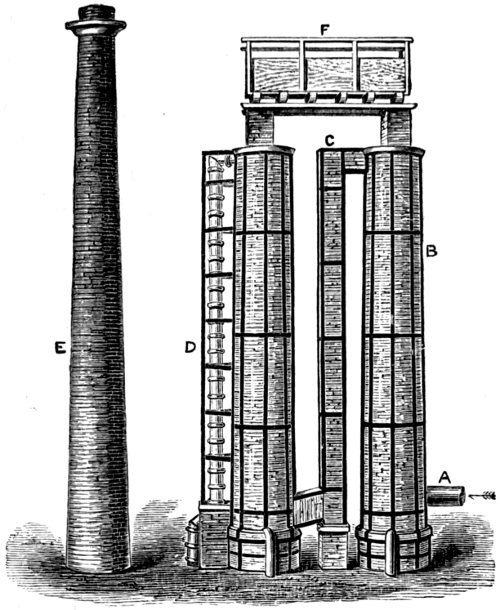
Fig. 8. PREPARATION OF HYDROCHLORIC ACID
By another piece of legislation, which at first sight seems to be wholly unconnected with Chemistry, hydrochloric acid acquired a greatly enhanced value. In 1861, the tax on paper was removed, and in the next twenty years the demand for that commodity increased so much that raw material both cheaper and more 46 abundant than rag had to be found. Esparto grass and eventually wood pulp proved successful substitutes. There is really very little difference in composition between cotton and linen rag on the one hand and wood fibre on the other, for both are mainly composed of cellulose, which is a definite chemical compound. Wood fibre is the less pure, and it is also coloured, and therefore has to be bleached before it can be used for making white paper. It was this circumstance which led to the greatly increased demand for hydrochloric acid.
At the beginning of this chapter, it was mentioned, in passing, that hydrogen chloride gas is a compound of hydrogen and chlorine. The latter element is a very active bleaching agent, and is most easily obtained by treating hydrogen chloride or its solution in water with pyrolusite (black oxide of manganese), whereby the hydrogen is oxidized, forming water, and chlorine gas is set free. Being a gas, chlorine is not convenient to handle in large quantities; it is, therefore, converted into bleaching powder, commonly but wrongly called chloride of lime.
Bleaching Powder. The manufacture of bleaching powder is carried out in the following way. Slaked lime to the depth of 3 or 4 in. is spread over the floor of a special chamber which can be made gas-tight. The lime is raked up into ridge and furrow, and the chamber is filled with chlorine. At the end of about twenty-four hours, the greater part of this chlorine will have been absorbed by the lime. The chamber is then opened, the lime is raked over to expose a fresh surface, and the process of chlorination is repeated. Generally this is sufficient; the bleaching powder should then contain about 35 per cent. of available chlorine.
The demand for bleaching powder is great and steadily increasing. The price of 35 per cent. bleaching powder 47 has never been less than about £5 a ton,[3] so that it is perhaps unnecessary to add that the absorption of hydrogen chloride gas is now made so complete that it is well within the requirements of the second Alkali Act.
Chlorides. The salts of hydrochloric acid are called chlorides, and the most important of these is sodium chloride or common salt—a body that is so well known that it need not be described here.
Although the quantity of this substance required for domestic purposes is very large, it is, nevertheless, small by comparison with that which is used for industrial purposes. It has already been mentioned that salt is the starting-point for the manufacture of washing soda by the Leblanc process, and, in addition to this, it is employed in the glass industry to produce whiteness and transparency in certain kinds of glass; in pottery, for glazing earthenware; in soap-making, for salting out the crude soap; and in the dye trade as a mordant, and also for improving the quality of certain colours. A full account of the salt industry is given in another volume of this series.
Hydrofluoric Acid. When calcium fluoride (fluorspar, Derbyshire spar, or blue-john) is warmed with concentrated sulphuric acid in a leaden dish, hydrogen fluoride gas is evolved, and this, when dissolved in water, gives hydrofluoric acid.
The peculiar property of this substance is that it has a very marked corrosive action on glass. It cannot, therefore, be kept in glass vessels, but must be stored in bottles made of hardened caoutchouc. On the other hand, it is this same property which gives it its place in commerce. As far back as 1670 it was used for etching on glass. The process is a very simple one. The article is first coated with wax, which is then removed in places by a sharp pointed tool. When 48 exposed to the action of the gas or its solution, corrosion takes place only where the glass has been laid bare, the other parts being protected by the wax. After a short interval, the wax can be melted off, and the design made more distinct by rubbing in some opaque cement. For general trade purposes, such as the stamping of lamp chimneys or electric light bulbs, a quicker method is required. In this case, a preparation of hydrofluoric acid which can be applied with a rubber stamp is used.
Fluorspar or calcium fluoride is the most important salt of hydrofluoric acid. It is a commonly occurring mineral, and besides its use for the preparation of the acid, it is employed in many metallurgical operations to form a fusible slag.
Hydrobromic and Hydriodic Acids are not much used, but their salts, the bromides and iodides respectively, are of great technical importance. Silver chloride, bromide, and iodide, are sensitive to light, and mixed with gelatine they form the emulsion which is spread over photographic plates and papers. Potassium bromide and iodide are also well known to photographers.
When the halogen salts of silver are exposed to light, an extremely subtle chemical change takes place, which is only made apparent when the plate or paper is developed. Then the silver salts on which the light has fallen are reduced to metallic silver, and this reduction is greatest where the light was most intense, and in other places is proportional to the light intensity. A very faint image may appear on the plate while it is in the developer, but generally the image is only brought out clearly when the plate, film, or paper is placed in “hypo” solution, which dissolves out the silver salts which have not been changed, leaving the metallic silver unaffected.
Carbon. When any product of animal or vegetable life is strongly heated in a vessel from which all air currents are excluded, a mixture of gases and liquids is driven off, and a charred mass remains. This residue, from whatever source obtained, is composed mainly of the element carbon. It sometimes happens that a loaf of bread or a cake is left in the oven and forgotten. In popular language it is then said to be “burnt to a cinder”; in reality, the surface layers have been converted into carbon.
Carbonic Acid. If carbon is heated in an open vessel provided with a good draught, it glows and in time disappears, because it combines with oxygen to form an invisible gas, carbon dioxide or carbonic acid gas, which, when dissolved in water, forms carbonic acid.
Compared with the acids which have been described in the foregoing chapters, this is a very feeble acid; it changes the colour of litmus to a wine red, not a bright pink; its taste is just pleasantly acid, and its solvent action on metals and limestone is very small indeed. The solution of the acid, obtained by passing carbon dioxide into water, is, of course, very dilute, and it cannot be concentrated by evaporation, since this only results in expelling the carbon dioxide from solution, leaving pure water.
Soda Water. In the case of most gases, the weight which dissolves in a given quantity of water is proportional to the pressure. This is true for carbonic 50 acid gas. Under a pressure of 4 atmospheres, the weight of gas which dissolves is four times as great as under a pressure of one atmosphere.
Soda water is water charged with carbon dioxide under pressure. This pressure is maintained from the time it leaves the manufacturer to the time it reaches the consumer by the strong walls of the syphon or bottle. Immediately this pressure is released, the greater part of the excess gas escapes, producing effervescence. It is, however, curious to note that all the gas which ought to escape when the pressure is reduced does not do so at once. If soda water is allowed to stand in an open glass until it becomes “flat,” a brisk effervescence can be started again by dropping a lump of sugar into the quiescent liquid. Soda water remains supersaturated with gas for some time after the pressure has been released.
Calcium Carbonate. The salts of carbonic acid are called carbonates. Calcium carbonate is one of the most abundant substances in Nature. The white cliffs of the east and south coasts of England, and those of France across the intervening sea, are the exposed parts of enormous beds of chalk or calcium carbonate. Whole mountain ranges in various parts of the world are composed of limestone, which in some cases is mainly calcium carbonate, and in others a mixture of this substance with magnesium carbonate. Marble, whether white, black, or variegated, is almost pure calcium carbonate, the differences of colour being due to insignificant traces of iron and other foreign matter. In Iceland spar and calc spar, sometimes called dog-tooth spar, we have two transparent crystalline forms of this same substance.
Connected with the animal kingdom there are forms of calcium carbonate no less varied in appearance. 51 Egg shells are composed of this substance, and so are oyster shells and the hard external coverings of some of the lower animals. The mother-of-pearl lining of the oyster shell, and also the pearl itself, are secretions of calcium carbonate. The beauty of the last-named variety is due to the external form and to minute inequalities of the surface, which cause the resolution of white light into colours seen in the spectrum or in the rainbow. The coral reefs or atolls of the Southern oceans, which may be miles in breadth and hundreds of miles in length, are all composed of calcium carbonate, which a tiny marine animal has formed for its own support and protection.
It is perhaps somewhat surprising at first to be told that all these forms are composed of the same chemical substance, yet on this point the evidence is definite and unmistakable. All the varieties dissolve readily in dilute hydrochloric acid with effervescence caused by the escape of carbon dioxide gas; moreover, if any of the purer forms, such as pearl, marble, or Iceland spar, are heated to redness for some time, they all lose about 44 per cent. by weight, leaving a residue which is pure lime.
Quicklime. The making of lime from limestone or chalk is called lime burning. The operation is carried out in a structure called a lime kiln, which is usually a barrel-shaped vertical shaft surrounded by substantial brickwork. There are two main methods of procedure, the one continuous and the other intermittent. In the continuous process, the kiln is filled up with limestone and fuel (generally coke) in alternate layers. Combustion is started at the bottom and maintained by a regulated draught. As the charge works down, the addition of limestone and fuel is continued from the top, while the lime is removed from the bottom of the 52 kiln. The lime produced by this method has the ashes of the fuel mixed with it. To avoid this, the more modern type of kiln has four lateral fire grates outside the actual kiln.
For the intermittent method, a kiln is required which has a fireplace at the bottom. Over this a rough arch is built of large pieces of limestone, laid dry, and then the kiln is filled up with pieces of limestone which decrease in size from below upwards. The fire is kindled beneath the arch and urged by a regulated draught. The heating is maintained for three days and nights, after which time the charge is allowed to cool down.
Carbonic Acid Gas in Nature. Although the solvent action of carbonic acid is very small compared with that of strong acids, it is nevertheless great in comparison with that of water. This is shown especially in its action on limestone, an action from which several important consequences arise. Rain, as it falls through the air, dissolves a little carbon dioxide and, although this is only an exceedingly dilute solution of a very weak acid, its cumulative effect, especially in limestone districts, is very great; it hollows out enormous caves and causes the formation of those fantastic creations in stone known as stalactites and stalagmites.
When a drop of water charged with carbonic acid gas falls on limestone, it dissolves a little of that substance, forming calcium bicarbonate, which may be regarded as a compound of calcium carbonate, carbon dioxide, and water. Little by little, the solid rock is hollowed out and a cave, or perhaps an underground watercourse, is formed.
Again, the drop of water charged with calcium bicarbonate may find its way to the roof of a cave. As it hangs from the roof while it gathers strength to 53 fall, a little of the carbon dioxide escapes, and a minute quantity of calcium carbonate is deposited. In this way, a stalactite looking like an icicle in stone gradually grows downwards.
When the drop reaches the floor of the cave, a little time elapses before it sinks into the ground; again a little carbon dioxide escapes, and a small quantity of calcium carbonate is formed. Little is added to little, and in the course of ages the stalagmite grows upward from the floor and ultimately meets the stalactite to form a continuous column of glistening crystallized calcium carbonate.
Hard and Soft Water. Water that is used for domestic or manufacturing purposes is described as either hard or soft. Soft water produces a soap lather almost at once; hard water forms at first a scum or curd which has no detergent properties, and only after a time gives the soap lather which is required. The difference is due to the relative amount of dissolved solid contained in the water.
Only distilled water or rain water collected in the open country is perfectly soft, for this is the only kind of water which on being evaporated to dryness leaves no solid residue. In districts where the underlying strata are composed of hard insoluble rock, such as granite or millstone grit, the water contains very little dissolved matter and is relatively soft. In a limestone or chalk country, water is very hard and in many cases has to be softened either before delivery or before use.
The chief impurities which cause hardness are the chlorides, sulphates, and bicarbonates of magnesium and calcium. The chlorides and sulphates are not affected in any way by boiling, and the hardness which is due to them is said to be “permanent.” The bicarbonates, 54 on the other hand, are decomposed when the water is boiled, and then they cease to cause the water to be hard. This part of the hardness is spoken of as “temporary” hardness.
Let us now consider what calcium bicarbonate is and how it is formed. It is a compound of calcium carbonate and carbonic acid, and is formed by the solvent action of carbonic acid on limestone or chalk. The compound is soluble in water; but when the solution is boiled, the carbonic acid is broken up, carbonic acid gas is expelled from the solution, and calcium carbonate is formed.
Temporary hardness is the more troublesome. In the first place, the bicarbonates, especially that of calcium, often form the greater part of the dissolved impurity. Moreover, when the water is boiled, although the hardness is removed, the insoluble calcium carbonate is a source of trouble, for it gradually settles down into the hard mass known as “fur” in kettles and “scale” in boilers.
It is perhaps necessary at this point to emphasize the fact that matter suspended in water does not make it hard, and it is only matter which is dissolved which makes any difference in this respect.
Since the softening of temporary hard water by boiling has the undesirable feature of introducing solid matter into the boiler, it is customary now to treat this water chemically. The following is the process most generally used. Quicklime or slaked lime is stirred into the water until the mixture gives a faint brown coloration when a drop of silver nitrate is added to a small test portion. Unsoftened water is then added until a sample just ceases to give this test. The temporary hardness has then been removed, and it is only necessary to allow the suspended matter to settle.
The explanation of the method is as follows. The 55 lime which is added neutralizes the carbonic acid combined with the calcium bicarbonate, and the result is the same as in the former case where this carbonic acid was decomposed by heating, for calcium carbonate is thrown out of solution.
For domestic purposes, water is softened by the addition of washing soda. Since this reacts with all the calcium and magnesium compounds forming the insoluble carbonates, all hardness, both temporary and permanent, is removed.
The acids which are grouped in this chapter are not in themselves of much interest, though some of their salts are extremely important compounds.
Bone. Much of the refuse bone, sooner or later, reaches the marine store, and from that point starts on a career of usefulness in the industrial world.
“Green bone,” as it is then called, may have fat adhering to it or confined in its hollow interior as marrow. This is recovered by treatment with benzine, and after that the bone is subjected to the action of superheated steam in order to convert cartilage into glue. In some cases, the residue is then ground up to make bone meal, which is valuable as a manure because of the calcium phosphate which it contains. In this way, the phosphate returns again to the animal kingdom, for it supplies plants with the phosphates that they require, and from the vegetable kingdom it passes to animals and helps to build up bone again.
Calcium Phosphate and Bone Black. Instead of being ground up, bone may be heated in a retort in much the same way as coal is treated for the manufacture of coal gas; bone oil is distilled off, and a non-volatile residue, called bone black or animal charcoal, remains. This contains about 90 per cent. of calcium phosphate and 10 per cent. of finely divided carbon disseminated throughout the mass. It has the peculiar property of absorbing colouring matter, and is used for this purpose in the sugar industry and in the preparation of fine chemicals.
Phosphoric Acid. After being some time in use, bone black loses the property of absorbing colouring matter; and though it can be “revived” several times by heating it strongly in a closed retort, it ultimately becomes spent and of no further use to the sugar refiner. It is then heated again, this time in an open vessel, until all the carbon is burnt away. The residue is now a greyish solid consisting mainly of calcium phosphate. This, supplemented with native phosphate, which is probably fossilized bone, is used for the preparation of phosphoric acid.
The salt is decomposed by sulphuric acid in wooden vats; calcium sulphate is formed, and ultimately settles on the bottom of the vat, leaving a clear supernatant liquid, which is a dilute solution of phosphoric acid. This liquid is drawn off and evaporated to a syrup. This is “syrupy” phosphoric acid. On being still more strongly heated, the syrup loses still more water, and a semi-transparent glassy-looking substance, called metaphosphoric acid, remains.
Superphosphate. All fertile soils, especially those on which wheat is to be grown, must contain a certain amount of phosphate. With this, as with all other plant foods, the actual percentage weight required in the soil is very small indeed, but it is necessary that it should be disseminated throughout the soil. Even distribution is very difficult to secure in the case of a substance like calcium phosphate, which is practically insoluble in water.
To get over this difficulty, calcium phosphate is converted into a mixture known as “superphosphate” by the following process. Bone ash or the mineral phosphate is finely ground and thoroughly mixed by machinery with two-thirds its weight of sulphuric acid from the lead chambers. After a time, this mixture sets to a 58 hard mass, containing principally gypsum and calcium tetrahydrogen phosphate. It is then ground up finely and is ready for use.
The special modification of calcium phosphate contained in superphosphate is soluble in water. It is, therefore, carried into the soil in solution, and in this way very evenly distributed. In the soil it reacts with the lime or chalk which is present, and is gradually reconverted into insoluble calcium phosphate.
The manufacture of superphosphate is a very important industry. The weight of the substance produced annually in Great Britain alone is not far below a million tons.
Basic Slag. In the Bessemer process for converting iron into steel, cast iron is melted up in a vessel called a converter and, by the aid of a powerful blast blown through the molten iron, most of the impurities are burnt off. If, however, phosphorus and sulphur are present, they are not removed if the converter has a silica (acid) lining. The original Bessemer process was, therefore, modified by Thomas and Gilchrist, and the converter for this kind of iron is lined with dolomite and lime (basic lining). Phosphorus is then converted into phosphate and retained by the lining, which is subsequently removed, ground up finely, and sold as “basic slag.”
Boric Acid, or boracic acid, is familiar because it is used in medicine as a mild antiseptic; it is also employed as a preservative for food. It is a white crystalline compound, sparingly soluble in water. It has no well-marked taste, and causes only a partial change in the colour of litmus solution; it is, therefore, one of the weak acids. It does not dissolve metals, but it displaces carbon dioxide from carbonates, forming salts.
Borax, the best known salt of boric acid, is used in laundry work and also for making some enamels, for when it is strongly heated it loses water, and ultimately melts down to a clear “glass” in which the oxides of metals will dissolve, yielding transparent substances which are beautifully coloured according to the nature of the oxide used. This property is often made use of in chemical analysis in what is known as the “borax-bead” test.

Fig. 9. BORIC ACID
Boric acid is a natural product; the method by which it is obtained is of some interest, because it is so simple, and because it shows how mere traces can be gradually accumulated until a very fair total is ultimately obtained. Moreover, the method is copied directly from Nature.
In the early years of the nineteenth century, certain jets of natural steam, called suffioni, which issue from the earth in Tuscany, were found to contain the vapour of boric acid. These jets of steam are of volcanic origin. The quantity of boric acid in the vapour is very small indeed; nevertheless, by the method which is adopted, it can be profitably recovered, and more than a ton of the solid is daily produced.
In the same country there are many lagoons, the water of which contains boric acid. It was rightly conjectured that this boric acid came from jets of steam which issued from the earth in the bed of the lagoon. This suggested the idea of building up an artificial lagoon around a group of jets.
Series of about five of these collecting basins (Fig. 9) are formed, each one at a slightly lower level than the one which precedes it. The first basin is filled with water from an adjacent spring, and this is allowed to remain for twenty-four hours. A sluice is then opened and the liquid contained in the first basin flows down to the second, where it remains for another day, and so on until it reaches the last basin of the series. The liquid by this time is almost fully charged with boric acid, but it contains only about 2 per cent., because the acid is so sparingly soluble in water.
From the last basin (A), the liquid runs into large vats (B, D), where the suspended impurities settle down. The solution of boric acid is then concentrated by causing it to flow over a broad inclined plane made of corrugated lead or through a series of shallow vessels heated by jets of natural steam. The hot liquid flows into another vat (C), and, as it cools, boric acid crystallizes out and is removed by perforated ladles.
The mother liquor from which the crystals have been withdrawn is, of course, a cold saturated solution of 61 the acid, and this is returned to the top of the incline to flow down again and lose more water. The boric acid is finally transferred to drying chambers, which are also heated by the natural steam.
Native borax or “tinkal” comes from Thibet and also from Ceylon. In California, a large quantity of borax is obtained from a borax lake, and also from the mud of marshes in its neighbourhood.
Silica. The element silicon does not occur in the free state in Nature, neither has any particular use been found for it, and therefore it is not often isolated except to provide a lecture specimen. The compounds of silicon, however, are both plentiful and important, especially silica, the oxide, and the silicates or salts of silicic acid.
The commonest forms of silica are sand, flint, and quartz. Silver sand is composed of small crystals of pure silica, while flint is the amorphous variety of the same substance. Quartz, or rock crystal, is a very hard and transparent mineral. It forms six-sided prisms ending in pyramids. It is distinguished from other common transparent minerals, such as calcspar, by the fact that it cannot be scratched even with a good knife or file, and that a drop of hydrochloric acid has no action on it. The melting point of silica is very high.
Sometimes silica is very delicately coloured with minute traces of metallic oxides and other substances, and these forms, because of their rarity and beauty, are more highly valued. Smoky quartz, cat’s-eye, and amethyst are some of the coloured varieties of quartz. Opal, agate, jasper, onyx, and chalcedony are, in the chemist’s classification, merely coloured flints.
In recent years, chemical apparatus has been made from pure fused silica. This can only be worked in the oxy-hydrogen blow-pipe flame or in the electric furnace; 62 nevertheless, crucibles, flasks, beakers, and retorts can be made. Silica ware has several advantages over glass, notably, that water has no action upon it at all; moreover, its coefficient of expansion is so very small that a piece of apparatus made of silica can be suddenly heated or cooled without risk of fracture; indeed, it can be made red-hot and cooled immediately by plunging into cold water.
Quartz or silica fibres, used for suspending magnets and other bodies in very delicate physical apparatus, are made in the following way. Molten silica is attached to the bolt of a crossbow, which is then released above a carpet of black velvet. As the bolt flies forward, it draws out the silica into a filament, which is so fine that it would be difficult to find were it not for the velvet background.
Silicic Acid itself is only of theoretical interest. It is obtained by adding hydrochloric acid to a solution of potassium or sodium silicate. It is a gelatinous substance of somewhat indefinite composition. It has no effect on litmus, no taste, and no solvent action; in fact, it is only recognizable as an acid because it dissolves in alkalis, forming salts called silicates. It is one of the weakest acids known.
The natural silicates are very abundant and varied; orthoclase or potash felspar, and albite or soda felspar, are those which most commonly occur. The former is potassium aluminium silicate, and the latter, sodium aluminium silicate. Iron is generally present in both as an impurity. The weathering of the felspars, in conjunction with the action of water, has produced the clays. In this way, pure white China clay has been formed from felspars which contain little or no iron, and the coarser kinds of clay from others containing a greater proportion of foreign substances.
Mica, which is used for making lamp chimneys, is a potassium aluminium silicate. Asbestos, meerschaum, beryl, garnet, jade, and hornblende are all silicates of various metals.
Glass is a complex mixture of insoluble silicates with excess of silica. The varieties in common use are soda glass, Bohemian glass, and lead glass (which is also called flint glass). Soda glass is mainly a mixture of calcium and sodium silicates, and is distinguished by its low melting point, which makes it easy to work at moderate temperatures. It appears in commerce as plate glass, window glass, and common bottles. Bohemian glass contains calcium and potassium silicates, and has a high melting point. It is used for making chemical apparatus. Lead or flint glass contains the silicates of lead and potassium; this is a dense glass, but at the same time rather soft. It takes a high polish and is used for making ornamental or cut-glass ware.
Remembering that glass is composed of the salts of silicic acid, the reader will readily understand that the mixture from which it is made must contain acidic and basic constituents. The acidic or acid-forming material is in every case silica or sand. This must be pure, and for all but the commonest kind of bottle or window glass, it must be free from iron, otherwise the glass will have a more or less pronounced greenish colour. It must also be fine and even grained. Formerly, the glass sands used in this country came from Holland and Belgium, but now supplies from several British sources are being successfully used.
The basic portion of the glass mixture differs according to the kind of glass required. An average mixture for soda glass contains sand, 20 parts; salt cake (sodium sulphate), 10 parts; quicklime, 5 parts; charcoal, 1 part. 64 For Bohemian glass, pearl ash (potassium carbonate) takes the place of salt cake, and no charcoal is necessary because the materials used are finer. For lead glass, the mixture is still further modified by the use of litharge, or more often red lead, in place of lime.
The ingredients are well mixed and thoroughly dried. Waste glass from a previous batch is also added. The mixture is heated to about 1200° C. in large pots made of Stourbridge clay, and the heating is continued for as much as sixteen hours, and until the whole of the material in the pot is molten and fairly mobile. Scum or glass-gall is removed, and when gas bubbles have disappeared, the temperature is allowed to fall to 700°-800°, when the glass becomes sufficiently viscous for subsequent working. The semi-fluid mass is then blown, moulded, or drawn, according to the kind of article that is required.
The physical properties of glass will now be considered in order that we may be able to account for its extended use. Such an inquiry as this, especially in the case of materials in common use, is often interesting, because it frequently happens that the special property upon which we set so much value is an abnormal one and, consequently, the feature which we take for granted is precisely the one into which we should inquire most closely.
The most striking feature of glass is its transparency. This property is abnormal, if glass is a solid. Consider what happens in most cases. A substance like nitre melts easily and in the molten state is perfectly transparent; when it cools, crystals form and, though these individually may be transparent, yet the solid mass is opaque. The reason for this is that the solid is not optically homogeneous, and therefore a ray of light cannot pass through it in a straight line. At each 65 facet of a crystal light is deviated and reflected, and in the end is almost wholly scattered. Consequently, an object, even if it can be seen at all, can be discerned only in a blurred and indistinct fashion through such a medium.
There are very good reasons, however, for supposing that glass is not a true solid but an extremely viscous liquid. If glass is heated, it softens and begins to flow very sluggishly at first, but afterwards more readily. There is no abrupt change, as there generally is in passing from the solid to the liquid state. Similarly in cooling, there is no point at which it is possible to say that the glass is solidifying. The view that this substance is really a liquid is perhaps a little startling at first, but it becomes less so when we observe that a long glass rod supported at its ends in a horizontal position sags in the middle and is permanently deformed.
To avoid that change which would be technically called solidification by a scientist, the article which has been fashioned in glass is cooled down very slowly and gradually. This part of the process is called annealing; it may occupy some days in extreme cases, and it points to the fact that experience has shown that it is necessary to guard against some change which would normally take place if this precaution were neglected.
The change in glass which annealing is intended to prevent is known as devitrification. In spite of all precautions, this does occur sometimes, and specimens of old window glass are often seen to have lost their transparency completely and to have an opalescent sheen. In these cases, the silicates have crystallized.
An extreme case of badly annealed glass is illustrated by Rupert’s drops, a scientific curiosity of very old standing. These are “tears” of glass made by dropping the molten substance into water. When the tail of 66 the drop is nipped off, the whole thing is shattered to powder with something like explosive violence. Clearly there is a very great internal strain, due to the fact that the outer parts have solidified and contracted, while the inner part is still warm and dilated.
Another remarkable feature of glass is the ease and simplicity with which it can be fashioned into articles of various shapes. As a plastic material, molten glass almost ranks with clay. This again is due to the property of passing through a viscous state, that is, one which is intermediate between a solid and a liquid.
Water Glass, or soluble glass, is mainly sodium silicate. It is made by fusing sand or powdered flint with caustic or with mild soda; sometimes, by digesting crushed flint or chert with caustic soda solution under considerable pressure in autoclaves or specially constructed boilers. In the latter case, no extraction is necessary; but in the former, the residue is treated with water and the solution evaporated until it becomes a viscous transparent liquid.
This liquid is used in various ways in industry. It is added to the cheaper varieties of yellow soap, and is employed as a mordant in dyeing and printing calico. An artificial sandstone is made by mixing sand, calcium chloride, and sodium silicate; the two last-named substances interact to form calcium silicate, which is insoluble in water. For domestic purposes, water glass is best known in connection with the preserving of eggs. When the film of water glass dries on the surface of the egg shell, the latter becomes impervious to air.
Organic Chemistry. About a century ago, when the science of Chemistry was still in its infancy, several substances were known which could then only be obtained from animals or plants. The composition of these substances was not understood, and they were not classified; moreover, since none of them had ever been prepared artificially, it was supposed that it was impossible to do this—the reason given was that “vital force” was necessary for their production. In time, however, some of the most typical animal and vegetable products were prepared in the laboratory, and the belief in vital force disappeared.
In later times it was proved that substances like sugar, starch, urea, indigo, and a great many more, all contain the element carbon. At the present time, more than 100,000 compounds of this element are known; and since they resemble one another, and at the same time differ in several important respects from the compounds of other elements, it is both natural and convenient that they should be classed together and studied separately. This branch of Chemistry is called organic. It must not, however, be supposed that all organic compounds are necessarily produced by some living organism. A great many are, but there are many more which are purely synthetic products.
Inorganic Chemistry includes all the other elements and their derivatives. The element carbon, and also some of its simpler compounds, such as carbon monoxide, carbon dioxide, carbonic acid, and carbonates, are more appropriately placed in the inorganic section.
The acids which have been considered up to this point are all inorganic acids, and those which follow are organic. Sulphuric, nitric, and hydrochloric acids are often distinguished as the mineral acids in contradistinction to oxalic, citric, tartaric, and some others which were first obtained from unripe fruits and therefore called vegetable acids.
Organic acids have all the general properties of the class, but they are much weaker than the mineral acids mentioned above. This is shown by their solvent action on metals, oxides, and carbonates, which is in all cases slight.
Vinegar is the trade name for what is essentially a dilute solution of acetic acid which has been made by the acetous fermentation of saccharine fluids containing weak alcohol. In addition to acetic acid, vinegar contains minute quantities of a large number of compounds. Some of these help to produce that agreeable flavour and aroma which distinguishes vinegar from diluted acetic acid. The nature and quantity of the flavouring constituents depend mainly upon the nature of the alcoholic solution used.
Since the acetic acid in vinegar is always produced by fermentation, all processes for the manufacture of vinegar are essentially arrangements for promoting the vigorous growth and development of Mycoderma aceti, the organism which produces the vinegar ferment.
Like all other plants, Mycoderma aceti will flourish only under certain favourable conditions. In the first place, it requires nourishment, and therefore certain nitrogen compounds and salts must be present in the alcoholic solution. These are contained in wines, beer, cider, and malt liquors, but not in spirits of wine, which is pure alcohol distilled from liquids which have undergone vinous fermentation. If the plant is placed in 69 dilute spirits of wine, only a very little acetic acid is formed, and then the action ceases because the solution does not contain the necessary food substances. Temperature also has a very marked effect on growth, the most favourable range being between 68° and 95° F.
Alcohol is changed to acetic acid by the process of oxidation, and therefore, in making vinegar, arrangements have to be made to bring together weak alcohol and air in the presence of the plant. The ferment which is secreted by the plant then causes an acceleration of the reaction. There is a considerable amount of similarity between fermentation and contact action. In this connection, it is interesting to note that the conversion of alcohol into acetic acid can also be brought about by exposing a mixture of alcohol vapour and air to the action of platinum black; in fact, there is one process for making vinegar in this way.
French Vinegar. New wine, especially that which contains a low percentage of alcohol, is liable to many kinds of “sickness.” It may turn bitter, it may turn sour, or it may undergo what is called lactic fermentation. In either case, it becomes unsaleable as a beverage. Wine which has turned sour is the best material for making vinegar, and when this is done by the French or slow process, a product with a very fine bouquet is obtained.
The methods adopted are very simple. Formerly, the wine was poured into barrels leaving the top portion empty, and providing for a current of air over the surface. The barrels were often set up in rows in the open air in an enclosure which was then known as a “vinegar field.” The process of souring which had already begun went on naturally, and in the course of a few months, nearly the whole of the alcohol was converted into acetic acid.
The process now in use in some of the French factories is somewhat similar. Large casks holding about 100 gallons are set up in a room, and provision is made for keeping the temperature uniform. Each cask is first acidulated by allowing strong vinegar to stand in it until the vinegar plant has developed on the surface. The casks are then filled up very gradually by adding a few gallons of wine every eight or ten days. When the cask is full, a fraction of the contents is drawn off and replaced by wine. The process then becomes continuous, until it is necessary to clean out the generator and start again.
In recent times, the manufacture of wine vinegar has been carried out on more scientific principles. The vinegar plant is actually cultivated and examined microscopically before being used, in order to make sure of the absence of moulds and bacteria, which set up other fermentations, producing substances which affect adversely the taste and aroma of the finished product. The cultivated ferment is then added to the wine in shallow vessels and the process is carried on as described above.
Malt Vinegar. A dilute solution of alcohol which is made from malt by fermentation with yeast contains the nutritive substances necessary for the growth of the vinegar plant, and can therefore be used as a starting-point for the manufacture of vinegar. Sprouted barley or malt is mixed with oats, barley, rice, or other starch-containing material. The mixture is mashed with warm water and then fermented with yeast, giving what is called “raw spirit.” This is converted into vinegar by the “quick” process.
The vinegar generator (Fig. 10) is a large barrel divided into three compartments by two perforated partitions. The lower disc is fixed about one-third of 71 the way up the barrel, and near it holes are bored to admit air. The upper disc, fixed near the top of the barrel, is perforated with a large number of small holes which are partially stopped up with short threads or wicks, which hang from the under side. The space between the two discs is packed with specially prepared beech shavings, which have been left to stand in strong vinegar until they are covered with the vinegar plant.
Fig. 10. QUICK VINEGAR PROCESS
The weak spirit is delivered into the upper portion of the barrel and is distributed in very small drops by the threads; it then passes slowly over the vinegar plant, to which the air also has free access. When it reaches the bottom, it overflows into a reservoir and is again passed through the generator; this is repeated until the product contains the desired amount of acetic acid.
The principle of the quick vinegar process is the same 72 as that employed in making wine vinegar. The speed of the reaction is, however, greatly increased by having the ferment spread over a very large surface and by the free circulation of air. It is possible to make wine vinegar by the quick process, but it is not done, because the product is inferior in taste and aroma to that made by the slow process.
Both wine vinegar and malt vinegar when freshly prepared have a stupefying and unpleasant odour. Before the product is ready for the market, it has to be matured in barrels. During this process, a small quantity of alcohol which still remains in the vinegar combines slowly with some of the acetic acid, producing acetic ester, a substance which has a pleasant fruity odour.
The colour of wine vinegar is natural, but vinegar which is produced by the quick process is colourless or only faintly coloured. Since the public has a preference for vinegar which is brown in colour, the product of the quick process is coloured artificially, either by adding caramel or by preparing the weak spirit from malt which has been slightly charred in drying.
Industrial Acetic Acid. The solutions of acetic acid dealt with above would be too dilute for any industrial purpose; moreover, the acid can be obtained much more cheaply by the distillation of wood. When wood is subjected to a high temperature, it is converted into charcoal and, at the same time, an inflammable gas, an acid liquid, and tar are given off, and can be collected in suitable vessels. The following table, on page 73, gives the relative amounts of the various substances obtained from wood by dry distillation. The quantities are those derived from one cord, that is, 125 cu. ft.
| Charcoal in bushels. | Alcohol in gallons. | Calcium acetate in lbs. | Tar in gallons. | Wood oil in gallons. | Turpentine gallons. | |
| Hard woods | 40-50 | 8-12 | 150-200 | 8-20 | ||
| Resinous woods | 25-40 | 2-4 | 50-100 | 30-60 | 30-60 | Heavy woods 12-25 |
| Light woods 2-10 | ||||||
| Sawdust | 25-35 | 2-4 | 45-75 |
The aqueous liquid that distils over contains methyl alcohol (wood spirit), acetone, and acetic acid. The crude mixture is known as pyroligneous acid. This is neutralized with milk of lime or soda ash, which converts acetic acid into calcium or sodium acetate, but has no action on the methyl alcohol and acetone which are also present. The mixture is then distilled, when methyl alcohol, acetone, and water pass over into the distillate, leaving the acetate in the retort.
To obtain the free acid from the acetate, the latter is well dried and then distilled with concentrated sulphuric acid. Acetic acid, being the more volatile of the two acids, distils over, and is nearly pure.
The method of removing the last traces of water depends upon the fact that acetic acid solidifies at 17° C. The acid, which is nearly, but not quite, free from water, is cooled until a portion solidifies. The part which still remains liquid is poured away, and the process is repeated until a residue is obtained which solidifies as a whole. This is glacial acetic acid, so called because it is a mass of glistening plates which look like newly-formed ice.
Aluminium Acetate, made by dissolving alumina in acetic acid, is the “red liquor” which is used as a mordant in dyeing. It is a colourless liquid, but is called “red liquor” because it is used with dyes which give a red colour.
Ferrous Acetate, made in a similar way from scrap iron and acetic acid, is the “black liquor” used in dyeing.
Verdigris, or basic copper acetate, is a valuable pigment. It is made by interposing cloths soaked in vinegar between plates of copper. After the action has been allowed to go on for a long time, the plates are washed with water and the verdigris is scraped off. The finest verdigris is made in France in the wine-producing district around Montpellier. Here, instead of cloths soaked in vinegar, the solid residue from the wine presses is spread in layers between the copper plates. The product made in this way is called vert de Montpellier.
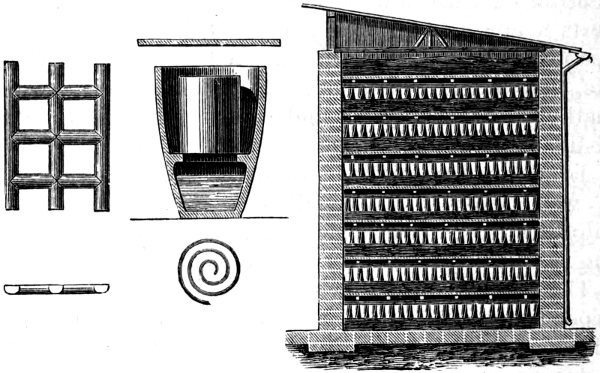
Fig. 11. DUTCH PROCESS FOR WHITE LEAD
Verdigris, like all the copper compounds, is extremely poisonous. It is very liable to be formed on the surface of copper utensils used for cooking purposes.
Lead Acetate, or sugar of lead, is used in large quantities in the colour industry for making various 75 reds and yellows. It is prepared by dissolving the metal or its oxide (litharge) in acetic acid.
The slow action which acetic acid vapour has upon the metal lead finds a very interesting application in what is known as the Dutch process for the manufacture of white lead[4] for paint. The metal is cast into grids or spirals, which are placed on the shoulders of the specially made pots sketched in Fig. 11. A little dilute acetic acid is poured into each of the pots, which are then arranged side by side on a thick layer of tan bark, stable manure, or other material which will heat by fermentation. The first layer of pots is then boarded over; another layer of pots is placed upon this, and so on, tier upon tier, until the shed is quite full. The heat developed by the fermenting material vaporizes the acetic acid, and this vapour corrodes the lead, forming basic lead acetate. The carbon dioxide which is also produced during fermentation converts the acetate into the carbonate, which falls as a heavy white powder into the pots.
Future Supply of Acetic Acid. When all the operations involved in the production of acetic acid from wood, from the felling of the tree to the final separation of the glacial substance, are taken into consideration, it will be readily understood how it is that this acid has never been cheap when compared with other acids used on an equally large scale. In addition to this, the competition for wood for paper-making and for the very numerous cellulose industries is rapidly increasing. It is, therefore, not surprising to learn that chemists have turned their attention towards the discovery of newer and cheaper methods of making acetic acid.
Such a process seems to have been worked out in 76 Germany. The starting-point is acetylene gas made by the action of water on calcium carbide. When this gas is passed through sulphuric acid containing suspended mercuric oxide or dissolved mercury salt, the acetylene is oxidized first to aldehyde and then to acetic acid.
If this process should prove to be successful, it will form the starting-point of a new and important industry, for, apart from the large amount of acetic acid which is used in commerce, there is the production of the very important solvent known as acetone, which can be made from acetic acid by a very simple operation.
Tartaric Acid. Grape juice contains a large quantity of potassium hydrogen tartrate dissolved in it; when the liquid is fermented and alcohol is formed, this salt crystallizes out because it is not soluble in alcohol. After the new wine has been poured off, the salt is found as a brownish crystalline residue adhering to the sides of the vat. Also the salt goes on crystallizing after the wine is put into barrels, and forms an incrustation on the sides. This is called the lees or sediment of wine. In commerce, the substance is known as argol (sometimes spelt argal), and also tartar of wine.
Crude argol is purified by dissolving it in water and destroying the colour by boiling with animal charcoal. When the clear liquid obtained from this mixture by filtration is evaporated, a white crystalline substance separates out. This is potassium hydrogen tartrate or cream of tartar.
Tartaric acid is obtained from cream of tartar. The salt is dissolved in water and nearly neutralized with milk of lime. Insoluble calcium tartrate is precipitated, and potassium tartrate remains in solution. A further quantity of calcium tartrate is obtained by adding 77 calcium chloride to the solution just mentioned. The two precipitates of calcium tartrate are then mixed and decomposed by dilute sulphuric acid, and after the calcium sulphate is filtered off, tartaric acid is obtained as a solid by evaporating the clear liquid.
The general properties of tartaric acid are well known. It is soluble in water, giving a solution which has a pleasantly acid taste.
Citric Acid. The sharp flavour of many unripe fruits is due to the presence of citric acid; the juice of lemons contains 5-6 per cent. of the acid. The free acid is obtained in a manner precisely similar in principle to that described for tartaric acid.
Oxalic Acid. Oxalic acid and its salts, the oxalates, are very widely distributed in the vegetable kingdom. These compounds are present in wood sorrel (Oxalis acetosella), in rhubarb, in dock, and in many other plants. The acid is made on a large scale by mixing pine sawdust to a stiff paste with a solution containing caustic soda and potash. The paste is spread out on iron plates and heated, care being taken not to heat the mixture to the point at which it chars. The mass is then allowed to cool, and is mixed with a small quantity of water to dissolve out the excess of alkali. This is recovered and used again.
Sodium oxalate, which is the main product of the reaction described above, is dissolved in water and treated with milk of lime, whereby insoluble calcium oxalate is obtained, which is subsequently decomposed with sulphuric acid, yielding oxalic acid.
Potassium hydrogen oxalate is sometimes called salts of sorrel, and potassium quadroxalate, salts of lemon. The most familiar use of the latter substance is in the removal of ink stains.
Oxalic acid and its salts are poisonous. The free 78 acid has sometimes been mistaken for sugar with fatal results.
Formic Acid (L. formica, an ant) is found both in the vegetable and in the animal kingdom. If the leaf of a stinging nettle is examined with a microscope, it is seen to be covered with long pointed hairs having a gland at the base. This gland contains formic acid. When the nettle is touched lightly, the fine point of the hair punctures the skin, and a subcutaneous injection of formic acid is made, which quickly raises a blister.
The inconvenience which arises from the stings of bees and wasps, also from the fluid ejected by ants when irritated, is due to formic acid. The remedy in each case is the same; the acid must be neutralized as quickly as possible with mild alkali, such as washing soda.
Formic acid was first made by distilling an infusion of red ants. It is now made from glycerine and oxalic acid.
The Fatty Acids. Animal fats and vegetable oils are similarly constituted bodies. They are composed mainly of three chemical compounds known as stearin, palmitin, and oleïn. Of these, stearin and palmitin are solids at ordinary temperatures, while oleïn is a liquid. Hard fats like those of mutton and beef are composed mainly of stearin; fats of medium hardness contain stearin, palmitin, and some oleïn; while oils such as cod-liver oil and olive oil are nearly pure oleïn.
Stearin, palmitin, and oleïn are analogous in composition to salts. Their proximate constituents are glycerine and certain organic acids, stearic, palmitic, and oleïc respectively.
In order to obtain the fat free from tissue which it contains in its natural state, it is tied up in a muslin bag and heated in boiling water. The fat is squeezed out through the meshes of the fabric and floats on the 79 surface of the water as an oil which solidifies on cooling. This clarified fat is called tallow.
All fats and vegetable oils can be resolved into their two constituents, the acid and the glycerine. This can be brought about by heating the fat with water to about 200° C. This operation must be carried out in a vessel capable of withstanding pressure and closed with a safety valve; otherwise, the requisite temperature could not be obtained. After this treatment, there is left in the vessel an oily layer which solidifies on cooling and an aqueous layer which contains the glycerine. The solidified oily layer is the fatty acid. In the case of mutton or beef tallow, it would be mainly a mixture of stearic and palmitic acids. This mixture is used to make “stearin” candles. The acids themselves are wax-like solids without any distinctive taste. Stearic acid melts at 69° C. and palmitic at 62° C. They have no perceptible action on the colour of litmus, neither have they any solvent action on metals or carbonates. We should not recognize these substances as acids at all were it not for the fact that they combine with alkalis, forming salts.
The salts of the fatty acids are called soaps. To make soap, the fat is boiled with caustic alkali or caustic lye, as it is more often called. This breaks the fat up primarily into the acid and glycerine; but in this case, instead of obtaining the acid as the final product as we did above by heating with water under pressure, we get the sodium or potassium salt of the acid according to the alkali used. When caustic soda is used, the product is a hard soap; when caustic potash is used, it is a soft soap. The treatment of fats in this way with caustic alkalis is called “saponification.”
Caustic and Mild. There are two classes of alkalis distinguished by the terms caustic and mild. If a piece of all-wool material is boiled with a solution of caustic soda or potash, it dissolves completely, giving a yellow solution. Mild alkali will not dissolve flannel, though it may have some slight chemical action causing shrinkage. Partly for this reason, and partly because commercial washing soda often contains a little caustic soda, woollen garments must not be boiled or even washed in hot soda water.
The disintegrating action of the caustic alkalis is also illustrated by the use of caustic soda in the preparation of wood pulp for paper making. Tree trunks are first torn up and shredded by machinery; but notwithstanding the power of modern machinery, the fibre is not nearly fine enough for the purpose until it has been “beaten” with a solution of caustic soda, whereby the pulp is brought to a smooth and uniform consistency like that of thin cream.
Mild Soda and Potash. Until the middle of the eighteenth century, it was thought that the soluble matter extracted from the ashes of all plants was the same. In 1752 it was shown that the substance obtained in this way from plants which grew in or near the sea differed from that from land vegetation by producing a golden yellow colour when introduced into the non-luminous flame of a spirit lamp, while that from land plants gave to the flame a pale lilac tinge. The former substance is now known as mild soda, and the latter as mild potash.
At this point it is well to make it clear to the reader that there are two bodies commonly called soda, and two called potash. One of each pair is caustic and one mild.
By a simple chemical test it is easy to distinguish a mild from a caustic alkali. When a little dilute acid is added to the former, there is a vigorous effervescence caused by the escape of carbon dioxide, but no gas is given off when a caustic alkali is treated in the same way. The liberation of carbon dioxide on the addition of acids shows that the mild alkalis are carbonates.
Washing Soda is so well known, that very little description of its external characteristics is necessary. It is a crystalline substance, easily soluble in water. The crystals, when freshly prepared, are semi-transparent; but after exposure to air for some time, they are found to lose their transparency and to become coated with an opaque white solid which crumbles easily. This change in appearance is accompanied by a loss in weight.
Crystals of soda melt very easily on the application of heat and, on continued heating, the liquid seems to boil. When this operation is carried out in a vessel attached to a condenser, the vapour that is given off from the melted soda condenses to a clear colourless liquid which, on examination, proves to be water. When no more water collects in the receiver, the vessel contains a dry, white solid, which by any chemical test that may be applied is shown to be the same as washing soda, but it contains no water of crystallization and has a different crystalline form. This substance is anhydrous sodium carbonate, or soda ash as it is called in commerce. When soda ash is mixed with water, it combines with about twice its own weight of that liquid, forming soda crystals again.
Washing soda, then, contains nearly two-thirds of its weight of water. Some of this water is given off spontaneously when the soda is exposed to air; the water may even be said to evaporate. This accounts for the loss of weight observed and also for the formation of the white layer of partially dehydrated soda over the surface of the crystal. The property of losing water in this way is common to most crystals containing a high percentage of water of crystallization. The phenomenon is known as “efflorescence.” It may here be observed that crystals of washing soda which have become coated over in this way contain relatively more soda than those which are transparent.
Natural Soda. In Egypt, Thibet, and Utah, there are tracts of country where the soil is so impregnated with soda that the land is desert. The separation of the soda from the earth is a simple operation, for it is only necessary to agitate the soil with water and, after the insoluble matter has settled down, to evaporate the clear solution until the soda crystallizes out.
In addition to alkali deserts, there are also alkali lakes. Those in Egypt are small, nevertheless, about 30,000 tons of soda per annum are exported from Alexandria. Owens Lake in California is said to contain sufficient soda to supply the needs of North America; while in the East African Protectorate, beneath the shallow waters of Lake Magadi (discovered in 1910), there is a deposit of soda estimated at 200,000,000 tons.
The Leblanc Process. At the present time, the greater part of the world’s supply of soda is made from common salt by two processes. The older of these, which is known as the Leblanc process, was introduced in France towards the end of the eighteenth century. In those days soda was very dear, for the main supply came from the ashes of seaweeds; wherefore the French Academy of Sciences, in 1775, offered a prize for the most suitable method of converting salt into soda on a manufacturing scale. The prize was won by Nicholas Leblanc, who in 1791 started the first soda factory near Paris. These were the days of the French Revolution; the “Comité de Sûreté Général” abolished monopolies and ordered citizen Leblanc to publish the details of his process.
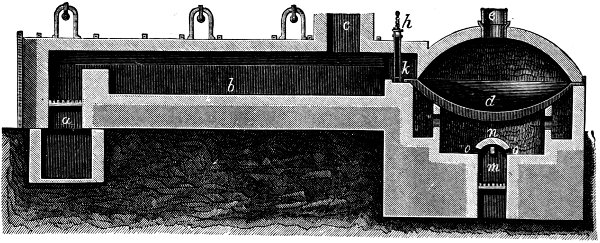
Fig. 12. SALT CAKE FURNACE
The first alkali works were established in Great Britain in 1814. The total amount of soda now made in this country every year is about 1,000,000 tons, of which nearly one-half is still made by the Leblanc process.
Salt Cake. The first stage of the Leblanc process consists in mixing a charge of salt weighing some hundredweights with the requisite amount of “chamber” sulphuric acid. The operation is carried out in a circular cast-iron pan (D, Fig. 12) about 9 ft. in diameter and 2 ft. deep. The pan is covered over with a dome of brickwork, leaving a central flue (E) for the escape of hydrochloric acid gas which is produced. At first, the reaction takes place without the application of heat, but towards the end the mass is heated for about one hour. The contents of the pan are then raked out on to the hearth of a reverberatory furnace (a, b) and more strongly heated. More hydrochloric acid gas is given off, and the reaction is completed. The solid product which remains is impure Glauber’s salt (sodium sulphate), and is known in the trade as “salt cake.”
Black Ash. In the second stage of the Leblanc process, salt cake is converted into black ash. The salt cake is crushed and mixed with an equal weight of powdered limestone or chalk and half its weight of coal dust. This mixture is introduced into a reverberatory furnace (Fig. 13) by the hopper K, and heated to about 1000° C. by flames and hot gases from a fire at a. During this operation, the mass is kept well mixed, and after some time it is transferred to h where the temperature is higher. The mixture then becomes semi-fluid and carbon monoxide gas is given off.

Fig. 13. BLACK ASH FURNACE
The formation of carbon monoxide within the semi-solid mass renders it porous. This is an advantage, because it greatly facilitates the subsequent operation of dissolving out the soluble sodium carbonate. The appearance of the flames of carbon monoxide at the surface of the black ash indicates the end of the process. The product is then worked up into balls and removed from the furnace.
The chemical changes which take place in making black ash are probably as follows: Carbon (coal dust) removes oxygen from sodium sulphate, which is thus changed to sodium sulphide. This substance then reacts with the limestone (calcium carbonate), forming sodium carbonate (soda) and calcium sulphide.
Extraction of Soda. It now only remains to dissolve out the soda from the insoluble impurities with which it is mixed in the black ash. It is evident that the smaller the amount of water used for this purpose the better, because the water has subsequently to be got rid of by evaporation. The process of extraction is, therefore, carried out systematically. The black ash is treated with water in a series of tanks which are fitted with perforated false bottoms. The soda solution, which is heavier than water, tends to sink to the bottom and, after passing through the perforations, is carried away by a pipe to the second tank, and so on throughout the series. The fresh water is brought first into contact with the black ash from which nearly all the soda has been extracted.
The method of finishing off the black ash liquor differs 87 according to the final product which the manufacturer desires to obtain, for the liquor contains caustic soda as well as mild soda. For the present, we will suppose that the end product is to be washing soda. In this case, carbon dioxide is passed into the liquor to convert what caustic soda there is into mild soda.
The clarified soda liquor is then evaporated until crystals of soda separate out. The first part of this process is carried out in large shallow pans (P. Fig. 13), using the waste heat of the black ash furnace, and finally in vats containing steam-heated coils. As the crystals separate out, they are removed, drained, and dried.
Alkali Waste. Black ash contains less than half its weight of soda, so that for every ton of soda produced there is from a ton and a half to two tons of an insoluble residue which collects in the lixiviating and settling tanks. This residue is known as alkali waste.
Alkali waste is of no particular value. It is not even suitable as a dressing for the land, and since it is not soluble in water there is no convenient means of disposing of it. Consequently, it is just accumulated at the works and, as the heap grows at an alarming rate, it cumbers much valuable ground. Moreover, it contains sulphides from which, under the influence of air and moisture, sulphuretted hydrogen is liberated. Alkali waste, therefore, has a very unpleasant odour.
The whole of the sulphur which was contained in the sulphuric acid used in the first stage of the process remains in the alkali waste, mainly as calcium sulphide. A plant for the recovery of this sulphur is established in some of the larger works. The alkali waste is mixed with water to the consistency of a thin cream, in tall, vertical cylinders. Carbon dioxide under pressure is forced into the mixture, and this converts the calcium 88 sulphide into calcium carbonate and sets free hydrogen sulphide, which, when burnt with a limited supply of air, yields sulphur.
By this process, the most unpleasant feature of alkali waste, namely, the smell, is removed. The calcium carbonate which remains is of very little value. Some of it is used in making up fresh charges for the black ash process and some for preparing Portland cement, for which finely-ground calcium carbonate is required; the remainder is thrown on a heap.
Bicarbonate of Soda. Bicarbonate of soda can be easily distinguished from washing soda. It is a fine, white powder similar in appearance to the efflorescence on soda crystals. It does not contain any water of crystallization.
When bicarbonate of soda is heated, it does not melt, and, as far as its external appearance is concerned, it does not seem to undergo any change. If, however, suitable arrangements are made, water and carbon dioxide gas can be collected, and the sodium bicarbonate will be found to have lost 36·9 per cent. of its weight. The substance which remains is identical with that obtained by heating soda crystals, that is, anhydrous sodium carbonate. Sodium bicarbonate is, therefore, a compound of sodium carbonate and carbonic acid.
The most familiar use of this compound is indicated by its common names “baking-soda” and “bread-soda.” It is mixed with dough or other similar material in order to keep this from settling down to a hard solid mass in baking. The way in which bicarbonate of soda prevents this will be readily understood when it is remembered that an ounce of this substance liberates more than 2,300 cu. in. of carbon dioxide when it is heated. When the bicarbonate of soda is well mixed with the ingredients of the cake or loaf and disseminated throughout the mass, each particle will furnish (let us say) its bubble of gas. Since these cannot escape, a honey-combed structure is produced.
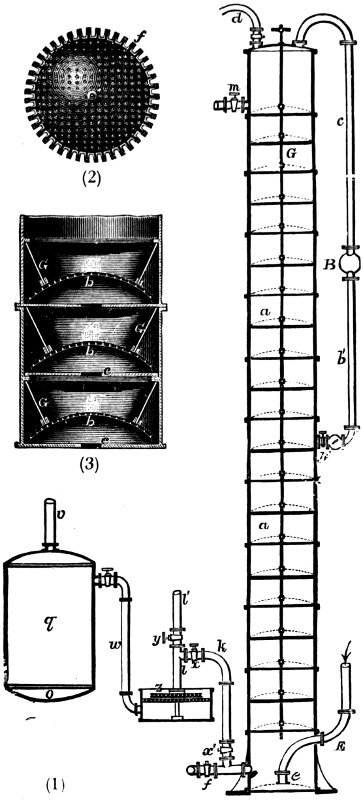
Fig. 14. THE SOLVAY PROCESS
Baking powder is a mixture of bicarbonate of soda and ground rice; the latter substance is merely a solid diluent.
The Solvay Process. Soda ash is one of the principal forms of mild alkali used in commerce. Large quantities of this substance are made by heating bicarbonate of soda. We shall now consider another alkali process in which this substance is the primary product.
For the greater part of the first century of its existence, the Leblanc soda process had no rival, although another method, known as the ammonia-soda process, was patented as early as 1838. In this case, however, as in many others, expectations based on the experiments carried out in the laboratory were not realized when the method came to be tried under manufacturing conditions. It was not until 1872 that Ernest Solvay, a Belgian chemist, had so far solved the difficulties, that a new start could be made. In that year, about 3,000 tons of soda were produced by the ammonia-soda or Solvay process, as it has now come to be known. Since then, however, the quantity produced annually has been steadily increasing, until at the present time it amounts to more than half of the world’s supply.
The Solvay process is very simple in theory. Purified brine is saturated first with ammonia gas and then with carbon dioxide. Water, ammonia, and carbon dioxide combine, forming ammonium bicarbonate, which reacts with salt (sodium chloride), producing sodium bicarbonate and ammonium chloride.
The principal reaction is carried out in a tower (Fig. 14 (1), a, a) from 50 to 65 ft. in height and about 91 6 ft. in diameter. At intervals of about 3½ ft. throughout its length, the tower is divided into sections by pairs of transverse discs, one flat with a large central hole, and one hemispherical and perforated with small holes (Fig. 14 (2)). The discs are kept in position by a guide rod G. Fig. 14 (3) shows a better arrangement of the guide rods. In modern works, the space between the discs is kept cool by pipes conveying running water. The ammoniated brine is led into the tower near its middle point. The carbon dioxide is forced in at E in the lowest segment, and as it passes up the tower it is broken up into small bubbles by the sieve plates. Sodium bicarbonate separates out as a fine powder, which makes its way to the bottom of the tower suspended in the liquid.
The perforated plates are necessary for the proper distribution of carbon dioxide through the brine. They are, however, a source of trouble, because the holes quickly become blocked up with sodium bicarbonate, and every ten days or so it is necessary to empty the tower and clean it out with steam or boiling water.
Recovery of Ammonia. The production of 1 ton of soda ash by the Solvay process involves the use of a quantity of ammonia which costs about eight times as much as the price realized by selling the soda. It is evident that the success of the process as a commercial venture depends largely on the completeness with which the ammonia can be recovered.
During the process, ammonia is converted into ammonium chloride, which remains dissolved in the residual liquor. From this ammonia gas is set free by adding quicklime and by blowing steam through the mixture. It is now claimed that 99 per cent. of the ammonia used in one operation is recovered.
Soda Ash. The bicarbonate of soda produced by the Solvay process is moderately pure. For all ordinary purposes, it is only necessary to wash it with cold water to remove unchanged salt, and after drying, it is ready to be placed on the market if it is to be sold as bicarbonate. The greater part of the Solvay product, however, is converted into soda ash by the application of heat. If soda crystals are required, the soda ash is dissolved in water and crystallized.
In many ways, the Solvay process compares very favourably with the older method. It is an advantage to start with brine, for that is the form in which salt is very often raised from the mines. The end product is relatively pure; moreover, it is quite free from caustic soda, which for some purposes for which soda ash is used is a great recommendation. There is no unpleasant smelling alkali waste. On the other hand, the efficiency of the Solvay process is not high, for only about one-third of the salt used is converted into soda. This would make the process impossible from the commercial point of view were it not for the cheapness of salt.
The Leblanc process, too, has its advantages. In the next chapter we shall see that it is adaptable for the production of caustic as well as mild alkali. The chlorine which is recovered in the Leblanc process is a very valuable by-product. In the Solvay process, chlorine is lost, for hitherto no practicable method has been found for its recovery from calcium chloride.
The position with regard to the future supply of alkali is very interesting. The competition between the Leblanc and the Solvay processes for supremacy in the market is very keen. At the same time, both processes are in some degree of danger of being supplanted by the newer electrical methods, which will be mentioned in the last chapter.
The following table shows very clearly the rapid progress made by the Solvay process in ten years. The quantities are given in tonnes (1 tonne = 0·9842 ton).
| 1884. | 1894. | |||
|---|---|---|---|---|
| Leblanc soda. | Solvay soda. | Leblanc soda. | Solvay soda. | |
| Great Britain | 380,000 | 52,000 | 340,000 | 181,000 |
| Germany | 56,500 | 44,000 | 40,000 | 210,000 |
| France | 70,000 | 57,000 | 20,000 | 150,000 |
| United States | — | 1,100 | 20,000 | 80,000 |
| Austria-Hungary | 39,000 | 1,000 | 20,000 | 75,000 |
| Russia | — | — | 10,000 | 50,000 |
| Belgium | — | 8,000 | 6,000 | 30,000 |
| 545,500 | 163,100 | 456,000 | 776,000 | |
Mild Potash. Potassium carbonate (mild potash) was formerly obtained from wood ashes. The clear aqueous extract was evaporated to dryness in iron pots, and the substance was on this account called potashes; later, potash. A whiter product was obtained by calcining the residue, and this was distinguished as pearl-ash. Chemically pure potassium carbonate was formerly obtained by igniting cream of tartar (potassium hydrogen tartrate) with an equal weight of nitre. It is for this reason that potassium carbonate is sometimes called “salt of tartar.”
About the middle of last century, natural deposits of potassium chloride were discovered in Germany. The beds of rock salt near Stassfurt are covered over with a layer of other salts, and for many years these were removed and cast aside as “waste salts” (abraumsalze). When at a later date they were examined more carefully, they were found to contain valuable potassium compounds, notably the chloride. After that discovery, 94 mild potash was made by the Leblanc process., and Germany controlled the world’s markets for all potassium compounds.
At the outbreak of war, the German supplies of potassium compounds ceased as far as the allied nations were concerned, and an older method of making potassium chloride from orthoclase or potash-felspar was revived. This involves the heating of the powdered mineral to a high temperature after mixing it with calcium chloride, lime, and a little fluorspar. The potassium chloride is then extracted from the fused mass with water. This method has been worked with great success in America, and it is claimed that potassium chloride can be made in that country at a cost which is lower than that formerly paid for the imported article.
Mild potash and soda are so very similar in chemical properties that in most cases it is immaterial which compound is used. In all cases in which there is this choice, soda is employed, both because it is cheaper and because it is more economical, for 106 parts of soda ash are equivalent to 138 parts of potash. There are, however, some occasions when soda cannot be substituted, notably for the manufacture of hard glass and soft soap, and for the preparation of caustic potash, potassium dichromate, and other potassium salts.
Potassium Bicarbonate. This resembles the corresponding sodium salt in nearly every respect. It is, however, much more readily soluble in water, so much so, that it is not possible to obtain this substance by the Solvay method. It is made from potassium carbonate by saturating a strong aqueous solution of that substance with carbon dioxide.
The Alkali Metals. The discovery of current electricity in 1790 furnished the chemist with a very powerful agency for bringing about the decomposition of compounds. Hydrogen and oxygen were soon obtained by passing an electric current through acidulated water; and in 1807, Sir Humphry Davy, who is perhaps better remembered for his invention of the miners’ lamp, isolated the metals sodium and potassium by subjecting caustic soda and caustic potash respectively to the action of the current.
Sodium and potassium are very remarkable metals. They are only a little harder than putty, and can easily be cut with a knife or moulded between the fingers. When exposed to the air, they rust or oxidize very rapidly, so much so that they have to be preserved in some mineral oil or in airtight tins. They are lighter than water, which they decompose with the liberation of hydrogen, and under favourable circumstances the hydrogen takes fire so that the metals appear to burn on the surface of the water. After the reaction is over and the sodium or potassium has disappeared, a clear colourless liquid remains which has a strongly alkaline reaction, and when this is evaporated until the residue solidifies on cooling, caustic soda or potash is obtained. For very special purposes, the caustic alkalis are sometimes made by the action of the metals on water, but for production on a large scale, less expensive methods are adopted.
Caustic Alkali is obtained from the corresponding 96 mild alkali in the following way. The substance—washing soda, for example—is dissolved in water and the solution is warmed. Lime is stirred into this solution, and from time to time a small test portion of the clear supernatant liquid is removed and mixed with a dilute mineral acid. When this ceases to cause effervescence, the change is complete. The clear liquid is now separated from the solid matter (excess of lime together with calcium carbonate) and evaporated in a metal dish. Since the caustic alkalis are extremely soluble in water, they do not crystallize as do most of the compounds previously described. Evaporation is, therefore, carried on until the liquid which remains solidifies when cold.
Caustic Soda. To describe the process by which caustic soda is manufactured, we must return to the making of black ash. The mixture from which black ash is made contains limestone. It is heated to 1000° C., which is a sufficiently high temperature to convert limestone into lime. When the black ash is subsequently treated with water, the lime which is present converts some of the mild alkali to caustic; consequently, black ash liquor always contains both alkalis.
When the manufacturer intends to make caustic soda and not soda crystals, the composition of the black ash mixture is varied by adding a larger proportion of limestone, so that there may be an excess of lime in the black ash produced. The treatment with water is carried out as described under washing soda, and then more lime is added to convert the mild soda into caustic soda. After the excess of lime and other suspended matter has settled down, the clear caustic liquor is evaporated in iron kettles until it becomes molten caustic, which will solidify on being allowed to cool.
There are various grades of caustic soda on the 97 market differing one from another in purity. The soap manufacturer uses caustic liquor or lye containing about 40 per cent. of caustic soda. For other purposes, the solid containing from 60 to 78 per cent. is used. Sometimes the product is whitened by blowing air through the strong caustic liquor or by the addition of a little potassium nitrate. Finally, for analytical purposes, caustic soda is purified by dissolving it in alcohol and subsequently evaporating the clear liquid.
Caustic Potash. The methods for the preparation of the corresponding potassium compound are precisely the same as those described for caustic soda; in fact, wherever the words sodium and soda occur in this chapter, the reader can always substitute potassium and potash respectively.
Caustic Lime. Apart from its use in making mortar and cement, lime is very often employed to neutralize acids. For this purpose, a suspension in water, called milk of lime, is generally used, for lime itself is not very soluble. Probably it is only the soluble part which reacts; nevertheless, as soon as this is used up, more of the solid dissolves, and in this way the action goes on as if all the lime were in solution.
Lime is also a very valuable substance in agriculture, especially on damp, boggy land, where there is much decaying vegetable matter, and on land which has been liberally manured. The soil in these cases is very likely to become acid and is then unproductive. Lime is added to “sweeten” the soil; in other words, to neutralize the acid.
Ammonia. The pungent smelling liquid popularly known as “spirits of hartshorn” is a solution of ammonia gas in water. It is a caustic alkali and, as such, is sometimes used to remove grease spots. Here, however, we shall consider ammonia only in connection 98 with ammonium salts, some of which are used in very large quantity as fertilizers.
The principal source of ammonia at the present time is the ammoniacal liquor obtained as a by-product in the manufacture of gas for heating and lighting. Coal contains about 1 per cent. of nitrogen, and when it is distilled, some of this nitrogen is given off as ammonia, which dissolves in the water produced at the same time. This liquid is condensed in the hydraulic main and in other parts of the plant where the gas is cooled down.
Gas liquor contains chiefly the carbonate, sulphide, sulpho-cyanide, and chloride of ammonia, together with many other substances, some of which are of a tarry nature. It would not be practicable to evaporate this liquid with a view to obtaining the ammonium salts, because it is only a very dilute solution. Hence, after the removal of tar, the liquor is treated in such a way that ammonia is set free.
In some cases the liberation of ammonia is accomplished by blowing superheated steam into the liquor, which sets free the ammonia which is combined as carbonate, sulphide, and sulpho-cyanide, but not that which is present as chloride. In other works, the gas liquor is mixed with milk of lime, which liberates all the combined ammonia. The ammonia is then expelled from the mixture by a current of steam or air and steam. In both cases, the gas which is given off is passed into sulphuric acid, whereby ammonium sulphate is formed in solution and afterwards obtained as a solid by evaporation.
Ammonium Chloride. Like all other alkalis, ammonia solution neutralizes acids, forming salts. With hydrochloric acid, it produces the white solid known as sal 99 ammoniac or ammonium chloride. This compound is familiar as the one required to make the liquid used in a Leclanché cell, which is generally used as the current generator for electric bells.
Ammonium Carbonate, which is also called stone ammonia and salt of hartshorn, is made by subliming a mixture containing two parts chalk and one part ammonium sulphate. It is a white solid which gives off ammonia slowly and is, therefore, used as the basis for smelling salts.
Ammonium Nitrate is obtained by passing ammonia gas into nitric acid until it is neutralized. It is a white solid, which melts easily on being heated, and breaks up into water and nitrous oxide (laughing gas), which is the “gas” administered by dentists. Ammonium nitrate is also used in the composition of some explosives: for example, “ammonite” is said to contain 80 per cent. of this substance.
Ammonium Sulphate is used chiefly as an artificial manure; the amount required for this purpose throughout the world is over 1,500,000 tons every year.
Synthetic Ammonia. Though the soluble compounds of nitrogen are fairly abundant, the supply is by no means equal to the demand, because such enormous quantities are required for agricultural purposes. It has been already said that ammonia is obtained as a by-product in the distillation of coal, and it has been repeatedly pointed out that our coal supplies are far from inexhaustible; moreover, coal gas may not always be used for lighting and heating. It, therefore, becomes a very important question as to how the future supply of ammonium salts is to be maintained.
Ammonia is a very simple compound formed from the elements nitrogen and hydrogen, and, as before mentioned, the supply of free nitrogen in the air is 100 literally inexhaustible. In recent years, the efforts of chemists have been directed towards finding a method of converting the free nitrogen of the air into some simple soluble compound. This problem is usually spoken of as the “fixation of nitrogen.”
In the Haber process, nitrogen obtained by the fractional distillation of liquid air is mixed with three times its volume of hydrogen, and this mixture is heated to between 500°C. and 700°C. under a pressure of 150 atmospheres (nearly 1 ton to the square inch) and in the presence of a contact agent. Under these conditions, nitrogen and hydrogen combine to form ammonia, which is condensed by passing the mixed gases into a vessel cooled with liquid air, any unchanged nitrogen and hydrogen being passed back again over the contact substance.
The problem of making ammonia from the air is closely connected with that of making nitric acid from the same source. In some experiments the two are combined, and ammonium nitrate is produced directly. Ammonia made by the Haber process, or some modification, is mixed with atmospheric oxygen and passed through platinum gauze heated to low redness. This results in the formation of nitric oxide, which is further oxidized by atmospheric oxygen; and finally, from a mixture of oxides of nitrogen, water vapour, and ammonia, synthetic ammonium nitrate is obtained.
One of the most noteworthy developments of modern chemical industry has been the increasing use of electricity as an agent for bringing about changes in matter. This has followed naturally from the reduction in the cost of electricity, due in great measure to the utilization of natural sources of energy which for untold ages had been allowed to run to waste.
This last achievement is likely to produce such a change in economic conditions that it is worth while giving a little thought to what may be called a newly-discovered asset of civilization. One example will make this clear. In the bed of the Niagara river, which flows from Lake Erie to Lake Ontario, there is a sudden drop of 167 ft. over which the water rushes with tremendous force and expends its energy in producing heat which cannot be utilized. This is a waste of energy, but it cannot be circumvented because no method has yet been found to control the waters of the Falls themselves. Nevertheless, by leading the head waters through suitable channels from the high level to the low, it is possible to use the energy to drive turbines, which, in their turn, drive dynamos which produce the current. This is merely the conversion of the energy of running water into electrical energy; and while the sun remains, this supply of energy will be forthcoming in undiminished quantity, because by the heat of the sun the water is lifted again as vapour, which descends as rain to replenish the sources from which the Niagara flows.
Electricity is employed in chemical industry in two ways. In the first place, it may be used to produce very high temperatures required for the reduction of some metallic ores, for melting highly-refractory substances, and for making steel. It is, however, rather with the second method, called electrolysis, that we are here mainly concerned.
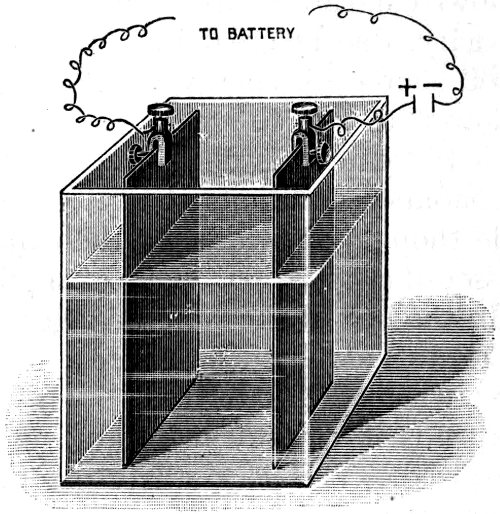
Fig. 15. THE ELECTROLYSIS OF SALT SOLUTION
Solutions of acids, bases, and salts, and in some cases the fused substances themselves, conduct the electric current; but at the same time they suffer decomposition. This method of decomposing a substance is known as electrolysis, or a breaking up by the agency of electricity.
The apparatus required in a very simple case is shown in Fig. 15. It merely consists of some suitable vessel to contain the liquid; two plates—one to lead the current into the solution, the other to lead it away again—and wires to connect the plates to the poles of a battery, 103 storage-cell, or dynamo. Each plate is called an electrode, and distinguished as positive or negative according as it is joined to the positive or negative pole of the current generator. By convention, electricity is supposed to “flow” from the positive pole of the battery to the positive electrode or anode, and then through the solution to the negative electrode or cathode, and so back to the negative pole of the generator, thus completing the circuit external to the battery.
When acids, alkalis, and salts are dissolved in water, there is strong evidence to show that they break up to a greater or less extent into at least two parts called ions. These are atoms, or groups of atoms, which have either acquired or lost one or more electrons.[5] They move about quite independently of one another and in any direction until the electrodes are placed in the liquid. Then they are constrained to move in two opposing streams—those which have acquired electrons all move towards the negative electrode, and those which have lost electrons towards the other. At the electrodes themselves, the former give up and the latter take up electrons, and become atoms again. Let us now consider a concrete example. Common salt is composed of atoms of sodium and atoms of chlorine paired. When a small quantity of this substance is dissolved in a large quantity of water, the pairing no longer obtains. The chlorine atoms move away independently accompanied by an extra satellite or electron, and the sodium atoms move away also but with their electron strength one below par. When the current is introduced into the liquid, the sodium ions travel towards the cathode and chlorine ions towards the anode, and when they reach the goal, sodium ions gain one electron and chlorine ions lose one, and both become atoms again. Chlorine atoms combine in pairs forming molecules and escape from the solution in the greenish yellow cloud that we call chlorine gas. The sodium atoms react immediately with water, forming caustic soda with the liberation of hydrogen.
To return now to practical considerations. The electrolysis of salt solution appears to be an ideally simple method of obtaining caustic soda and chlorine from sodium chloride. As a manufacturing process, it would seem to be perfect, for the salt is broken up directly into its elements and a secondary reaction gives caustic soda automatically. There is no “waste” as in the Leblanc process, and it does not require the use of any expensive intermediary substance afterwards to be recovered, as in the Solvay process. But, as very often happens when working on a large scale, difficulties arise, and these up to the present have only been partially overcome.
Some of the chlorine remains dissolved in the liquid and reacts with the caustic soda, forming other substances which, though valuable, are not easy to separate from the caustic soda. It is possible to get over this difficulty to some extent by placing a porous partition between the anode and the cathode, and in that way dividing the cell into cathodic and anodic compartments. As long as the partition is porous to liquids, it will allow the current to pass, but at the same time it will greatly retard the mixing of the contents of the two compartments. Porous partitions or cells which are in common use for batteries are made of “biscuit” or unglazed porcelain.
It must be remembered, however, that porous partitions only retard the mixing of liquids; they do not 105 prevent it. Moreover, a further difficulty arises from the fact that chlorine is a most active substance, and therefore it is difficult to find a material which will resist its corrosive action for any length of time, and the same difficulty arises in the case of the anode where the chlorine is given off.
Castner Process for Caustic Soda. The following is the most successful electrical process for the manufacture of caustic soda yet devised. It was introduced in 1892, and is known as the Castner process. It should be noted that the use of the porous partition has been avoided in a very ingenious way.
Fig. 16. THE CASTNER PROCESS
The cell (see Fig. 16) is a closed, rectangular-shaped tank divided into three compartments by two non-porous partitions fixed at one end to the top of the tank, while the other end is free and fits loosely into a channel running across the tank. The floor of the tank is covered with a layer of mercury of sufficient depth to seal the separate compartments. The two end compartments contain the brine in which are the carbon anodes; the middle compartment contains water or very dilute caustic soda in which the cast-iron cathode is immersed.
The current enters the end compartments by the carbon anodes and passes through the salt solution to the mercury layer which in these compartments are the cathodes. The current then passes through the mercury to the middle compartment, and then through the solution to the cathode, thence back to the dynamo. It is important to note that in the middle compartment the mercury becomes the anode.
Chlorine is liberated at the carbon electrodes, and when no more can dissolve in the liquid it escapes and is conveyed away by the pipe P. Sodium atoms are formed at the surface of the mercury cathodes in the outside compartments and dissolve instantly in the mercury, forming sodium amalgam.
While the current is passing, a slight rocking motion is given to the tank by the cam E. This is sufficient to cause the mercury containing the dissolved sodium to flow alternately into the middle compartment, and there the sodium amalgam comes into contact with water; the sodium is dissolved out of the mercury and caustic soda is formed. Water in a regulated stream is constantly admitted to the middle compartment, and a solution of caustic soda of about 20 per cent. strength overflows.
The production of caustic soda by an electrical method still remains to be fully developed. A process which gives only a 20 per cent. solution cannot be looked upon as final. In the meantime, other methods have been tried, in some of which fused salt is used in place of brine in order to give caustic soda in a more concentrated form. For a description of these methods, the reader must consult some of the larger works mentioned in the preface. Here we can only say that very great difficulties have been encountered, particularly in the construction of a satisfactory porous 107 diaphragm or, alternately, in devising methods in which this can be dispensed with.
Another interesting application of electrolysis is furnished by the use of copper sulphate in industry. When this salt is dissolved in water, it breaks up into copper ions (positive) and an equal number of negative ions, composed of 1 atom of sulphur and 4 atoms of oxygen (SO″4). Under the influence of the current copper ions travel to the cathode, and there by the gain of two electrons become copper atoms. Now, since copper is not soluble in copper sulphate solution, and is not volatile except at very high temperatures, it is deposited on the cathode in a perfectly even and continuous film when the strength of the current is suitably adjusted. This film continues to grow in thickness as long as the conditions for its deposition are maintained. If the current employed is not suitable, the metallic film is not coherent, and the copper may appear as a red powder at the bottom of the cell. Any other metal or impurity which might be present in the unrefined copper falls to the bottom of the tank.
Other metals are deposited electrolytically in exactly the same way. The metal to be deposited is joined to the positive pole and the article to be plated to the negative pole of the battery. Both are suspended in a solution of salt, generally the sulphate, of the metal which is to be deposited. Thus, for nickel plating, a piece of sheet nickel would be used in conjunction with a solution of sulphate of nickel or, better, a solution of nickel ammonium sulphate, made by crystallizing ammonium and nickel sulphates together. The current required is small; indeed, if it is too strong, the deposit adheres loosely to the article, and the result is, therefore, not satisfactory.
Electrotype blocks are also made by a similar process. 108 An impression of the article to be reproduced is made in wax, or some suitable plastic material, and polished with very fine graphite or black lead, in order to give a conducting surface. It is then suspended in a solution of copper sulphate and joined to the negative pole of the battery; a plate of copper connected with the positive pole is suspended in the same solution. When a weak current is passed, copper is deposited on the black-leaded surface and grows gradually in thickness, until at length it can be stripped off, giving a positive replica of the object.
THE END
Printed by Sir Isaac Pitman & Sons, Ltd. Bath, England
(v—1468c)