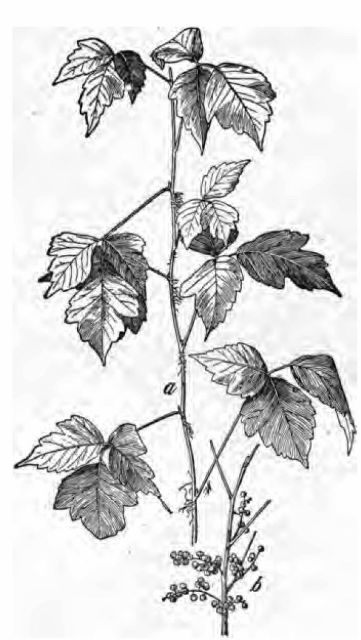
 Fig. 1.—Poison ivy (Rhus radicans or Rhus
toxicodendron). a, spray showing aerial rootlets and leaves; b,
fruit—both one-fourth natural size.
Fig. 1.—Poison ivy (Rhus radicans or Rhus
toxicodendron). a, spray showing aerial rootlets and leaves; b,
fruit—both one-fourth natural size.(Chesnut, Bulletin No. 20, Division of Botany, U. S. Department of Agriculture.)
The Project Gutenberg EBook of Some Constituents of the Poison Ivy Plant: (Rhus Toxicodendron), by William Anderson Syme This eBook is for the use of anyone anywhere at no cost and with almost no restrictions whatsoever. You may copy it, give it away or re-use it under the terms of the Project Gutenberg License included with this eBook or online at www.gutenberg.org Title: Some Constituents of the Poison Ivy Plant: (Rhus Toxicodendron) Author: William Anderson Syme Release Date: November 30, 2010 [EBook #34510] Language: English Character set encoding: ISO-8859-1 *** START OF THIS PROJECT GUTENBERG EBOOK SOME CONSTITUENTS--POISON IVY PLANT *** Produced by Bryan Ness, Josephine Paolucci and the Online Distributed Proofreading Team at https://www.pgdp.net. (This book was produced from scanned images of public domain material from the Google Print project.)
SUBMITTED TO THE BOARD OF UNIVERSITY STUDIES OF THE JOHNS HOPKINS UNIVERSITY IN CONFORMITY WITH THE REQUIREMENTS FOR THE DEGREE OF DOCTOR OF PHILOSOPHY
1906
1906
THE SUN JOB PRINTING OFFICE
BALTIMORE
Acknowledgments 4
Literature 5
Introduction 7
Work of Khittel 11
Work of Maisch 12
Work of Pfaff 13
Experimental 14
Gallic Acid 18
Fisetin 20
Rhamnose 23
The Poison 28
Potassium Permanganate as a Remedy for Rhus Poisoning 35
Summary 37
Biography 38
The author desires to avail himself of this opportunity to tender his thanks to those under whose guidance he has worked while a student at the Johns Hopkins University, namely to Professors Remsen, Morse, Jones, and Andrews, and to Doctors Acree and Tingle for instruction in lecture room and laboratory.
He is especially indebted to Dr. S. F. Acree, at whose suggestion this research work was undertaken, for counsel and assistance in its prosecution.
He would also thank Messrs. Parke, Davis and Co., of Detroit, Mich., for the preparation of the crude material used in this investigation, and the U. S. Department of Agriculture, Washington, D. C., for electrotypes of figures 17, 18, and 19 in Bulletin No. 20, Division of Botany.
Acides Gummiques, Garros (Dissertation) 1895.
American Chemical Journal.
American Journal of the Medical Sciences.
American Journal of Pharmacy.
Annalen der Chemie und der Pharmacie (Liebig).
Annales de Chimie et de Physique.
Berichte der deutschen chemischen Gesellschaft.
Biochemie der Pflanzen (Czapek) 1905.
Brooklyn Medical Journal.
Bulletin de la Société Chimique.
Bulletins 20 and 26 U. S. Department of Agriculture, Division of Botany.
Chemie der Zuckerarten, Von Lippmann, 1904.
Chemiker-Zeitung.
Comptes rendus.
Industries of Japan, J. J. Rein.
Journal of the Chemical Society.
Journal of Experimental Medicine.
Les Sucres, Maquenne, 1900.
Manual of Botany, 6th Edition, Gray.
Medical and Surgical Reporter.
New York Medical Record.
Proceedings of the American Pharmaceutical Association.
Treatise on Chemistry, Roscoe and Schorlemmer.
Über Mategerbstoff, Reuchlin (Dissertation) 1904.
Plants belonging to the natural order Anacardiaciæ (Cashew family or Sumach family) are found in all the temperate climates of the world and quite frequently in semi-tropical climates. Many of these plants play important parts in economic botany, yielding dye-stuffs, tanning material, wax, varnish, and drugs. Several species are poisonous. At least three poisonous species of the genus Rhus are found in the United States. These three are all common and well-known plants, but confusion frequently arises concerning them on account of the different names by which they are known in different localities. For example, poison ivy (Rhus toxicodendron or Rhus radicans) probably the best known poisonous plant in America, being found in all the States except those in the extreme West, is often confounded with and popularly called "poison oak." The true poison oak is the Rhus diversiloba of the Western States.[1] The third and most poisonous species of this plant is Rhus venenata or Rhus vernix; it is the Rhus vernicifera of Japan, from which Japanese lac is obtained. It is popularly known in the United States as "poison sumach," "poison dogwood" and "poison elder." It grows in swamps from Canada to Florida.
As the poison ivy is by far the most common of these plants in the Eastern States, a brief description of it is given here:[2] A shrub climbing by rootlets over rocks, etc., or ascending trees, or sometimes low and erect; leaflets 3, rhombic-ovate, mostly pointed, and rather downy beneath, variously notched, sinuate, or cut-lobed; high climbing plants (R. radicans) having usually more entire leaves. It is found in thickets, low grounds, etc. Greenish flowers appear in June.
Fig. 1.—Poison ivy (Rhus radicans or Rhus
toxicodendron). a, spray showing aerial rootlets and leaves; b,
fruit—both one-fourth natural size.In the general description of the order Anacardiaciæ, Gray[3] says: "Juice or exhalations often poisonous." Whether it is contact with some part of the plant, or with the exhalation from the plant, that causes the well-known skin eruption has been a topic for discussion ever since its source was known. On account of its intangible nature there has been more speculation than experimental evidence bearing on this question, although a few investigations have been made with the object of isolating the poison. It is most generally believed that the exhalations are poisonous. Dr. J. H. Hunt[4] states that the exhalations have been collected in a jar and found to be capable of inflaming and blistering the skin of an arm plunged into it.
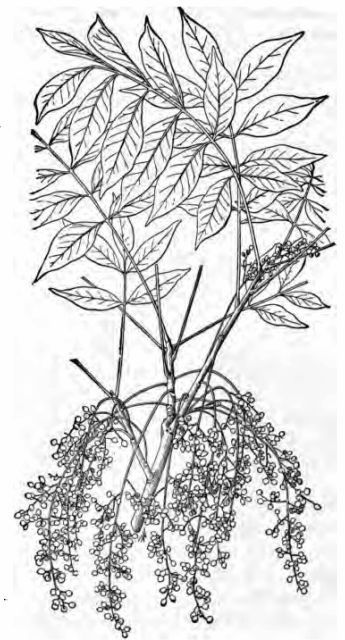 Fig. 3—Poison sumach (Rhus vernix), showing leaves,
fruit, and leaf-scars, one-fourth natural size.
Fig. 3—Poison sumach (Rhus vernix), showing leaves,
fruit, and leaf-scars, one-fourth natural size.Prof. J. J. Rein,[5] in his treatise on Lacquer Work, describes the poison of the Japanese lac tree, Rhus vernicifera, as being volatile, as do also the Japanese chemist Yoshida[6] and the French chemist Bertrand.[7] Recent work by Prof. A. B. Stevens,[8] however, seems to show that this poison is not volatile, and is similar to, if not identical with that obtained by Pfaff[9] from Rhus toxicodendron and Rhus venenata.
Not many cases of internal poisoning by Rhus toxicodendron are on record in medical literature. Two cases of poisoning from eating the fruit of this plant have been described.[10] The subjects of these cases were two children who had eaten nearly a pint of the fruit. The symptoms are described in detail, being in general, similar to those of alkaloidal poisoning. Warm water was given to promote emesis; afterwards large quantities of carbonate of soda were given in solution under the belief that it was an antidote to the poison. Otherwise they were treated on general principles. Both children recovered.
Another case of internal poisoning is the following:[11] Three children drank an infusion of the root of poison ivy thinking it was sassafras tea. The first of these cases was diagnosed as measles, but on the appearance of similar symptoms in the sisters of the first patient, the cause of the trouble was found. All recovered.
Dr. Pfaff[12] explains the few fatal cases that have followed Rhus poisoning on the assumption that enough of the poison was absorbed through the skin to cause renal complications in persons having chronic kidney trouble. He showed that the poison, when given internally, produces a marked effect on the kidneys, causing nephritis and fatty degeneration of this organ.
The irritating action of poison ivy has been attributed at different times to the "exhalation," to a volatile alkaloid, to a volatile acid, and to a non-volatile oil. Pfaff,[13] who made the most recent investigation of this poison, obtained from the plant a non-volatile oil having the same action on the skin as the plant itself. He found this oil in all parts of the plant and concluded that it was the active principle, and that one could be poisoned only by actual contact with some part of the plant. He assumed minute quantities of pollen dust to be in the air to account for the cases of "action at a distance" so frequently quoted. Pfaff says: "In my opinion, it is more than doubtful if ever a case of ivy poisoning has occurred without direct contact with the plant or with some article that has been in contact with the plant. The long latent period of the eruption in some cases may obviously render mistakes extremely easy as to the occasion when contact with the plant really occurred." Granting, however, that the active principle is practically non-volatile when isolated from the plant, we cannot say positively that it is not volatile in the[Pg 11] juices of the plant, or under the influence of vital forces. It is quite conceivable that the water transpired by the leaves of the plant may carry with it a quantity of the poison sufficient to produce the dermatitis on a person very susceptible to its action. It is also conceivable that a volatile poison manufactured by a living plant could become non-volatile by changes in it consequent upon the death of the plant.
Up to the present time, only three important chemical investigations of the active principle of Rhus toxicodendron have appeared in medical and chemical literature, these being the researches of Dr. J. Khittel, J. M. Maisch, a pharmacist, and Dr. Franz Pfaff, of the Harvard University Medical School, to whose work reference has been frequently made. The chemical work of these investigators and their conclusions are given here in some detail for the sake of completeness.
[1] Chesnut. Bull. No. 20, U. S. Dept. of Agr., Div. of Botany.
[2] Man. of Bot., p. 119.
[3] Man. of Bot., p. 119.
[4] Brook. Med. Jour., June, 1897.
[5] Rein, The Ind. of Jap., p. 338, et seq.
[6] H. Yoshida on Urushi Lacquer, Jour. Chem. Soc., 1883, p. 472.
[7] Ann. de Chem. et de Phys., Series VII, Vol. 12, p. 125, 1897.
[8] Amer. Jour. Pharm. 78, p. 53, Feb., 1906.
[9] An account of Pfaff's work will be found in another part of this paper.
[10] Amer. Jour. Med. Sci. 51 (1866), p. 560.
[11] Med. and Surg. Rep. 17, Nov., 1867.
[12] Jour. Exp. Med. 2 (1897), p. 181.
[13] Ibid.
The first attempt to find the poisonous constituent of this plant was made by Khittel in 1857. His work was published in Wittstein's Vierteljahrresschrift für praktische Pharmacie, VII, 348-359.[14] Khittel obtained 37-1/2 ounces of fresh leaves of poison ivy from the botanical garden in Munich, dried them, and got a residue of 9-1/2 ounces which he analyzed. Not detecting anything to which the poisonous qualities of the plant could be attributed, he made another series of experiments which, as he thought, showed that a volatile alkaloid is the poisonous constituent. It was obtained by the following process: "3 ounces of the powdered leaves were infused with hot distilled water, after three days strained, expressed, the liquid evaporated to 3 ounces, and with the addition of potassa, carefully distilled to one-half. The clear, colorless distillate had an alkaline reaction, and an odor resembling henbane or hemlock. It was saturated with sulphuric acid, evaporated, and treated with a mixture of equal quantities of alcohol and ether which left sulphate of ammonia behind, the solution was evaporated spontaneously, distilled with potassa, the alkaline distillate neutralized with hydrochloric acid, and a precipitate could now be obtained with chloride of platinum. Want of material prevented further experiments."
The editor of the American Journal of Pharmacy inserts the following note: "It would have been more satisfactory if the author had given some physiological evidence of the poisonous[Pg 12] nature of the alkaloid substance obtained. It is quite interesting to hear that the hitherto intangible venom of this plant has at last been detected."
[14] A free translation of this paper is given in Amer. Jour. Pharm. for 1858, p. 542.
The next investigation of this plant was made by Maisch in 1864. He criticizes Khittel's experiments as follows: "It is well known that the exhalations of Rhus toxicodendron exert a poisonous influence on the human body; the poisonous principle must, therefore, be volatile and, at the same time, be naturally in such a loose state of combination as to be continually eliminated and separated with the usual products of vegetable exhalations. It is natural to suppose that during the process of drying, the greatest portion of the poisonous principle should be lost. The loss must be still greater if the dried leaves are powdered, a hot infusion prepared from them, and this infusion evaporated down to the original weight of the dried leaves. It is obvious that Dr. Khittel could not have selected a better method for obtaining the least possible quantity of the poisonous principle, if, indeed, it could be obtained by this process at all."
Maisch then worked up 8-3/4 ounces of the leaves of the plant in a way to get the alkaloid, making some improvements on Khittel's method, but failed to find it. Believing that the poison was a volatile acid, he enclosed some fresh leaves of the plant in a tin box with several test papers. The blue litmus paper became red showing the presence of an acid. He concluded from this experiment that the exhalations of the leaves contained a volatile organic acid which he thought was the poisonous substance. To determine this point, he prepared the acid in larger quantity by macerating the leaves with water, expressing and distilling the expressed juice. He was poisoned in doing this work although he had not been affected by handling the living plant and had considered himself immune. He obtained an acid which investigation showed to be somewhat like formic acid, more like acetic acid, but having some reactions different from both. "Taking all the reactions together, it is unquestionably a new organic acid for which I propose the name of Toxicodendric Acid," writes Maisch. He further says: "That it is the principle to which poison oak owes its effects on the human system was proved to my entire satisfaction by the copious eruption and formation of numerous vesicles on the[Pg 13] back of my hand, on the fingers, wrists, and bare arms while I was distilling and operating with it. Several persons coming into the room while I was engaged with it were more or less poisoned by the vapours diffused in the room; and I even transferred the poisonous effects to some persons, merely by shaking hands with them.
"The diluted acid, as obtained by me, and stronger solutions of its salts, were applied to several persons, and eruptions were produced in several instances, probably by the former, though not always, which was most likely owing to the dilute state of the acid. Whenever this was boiled, I always felt the same itching sensation in the face, and on the bare arms, which I experience on continual exposure of my hands to the juice of the plant."
Toxicodendric acid was thought to be the active principle from the time of Maisch's work until the investigation by Pfaff in 1895.
[15] Proc. Amer. Pharm. Assn. 1865, p. 166, and Amer. Jour. Pharm. 1866, p. 4.
By far the most valuable work on Rhus toxicodendron is that of Pfaff. From a clinical study of Rhus poisoning, Pfaff came to the conclusion that the poison must be a non-volatile skin irritant. The more volatile the irritant, the quicker is its action on the skin. Formic acid acts very quickly; acetic acid, less volatile than formic, acts more slowly, but still much more quickly than poison ivy, the latent period of which is usually from two to five days. Pfaff thought that the volatile acid obtained by Maisch might have contained some of the poisonous principle as an impurity, but that it would not produce the dermatitis if prepared in a pure state. He therefore prepared a quantity of the acid by distilling the finely divided fresh plant with steam. The yield was increased by acidulating the mixture with sulphuric acid before the distillation. The acid distillate so obtained was freed from a non-poisonous oily substance by shaking the solution with ether. Barium and sodium salts were made by neutralizing the acid, and were purified by crystallization. Analysis showed them to be salts of acetic acid, and they gave the characteristic tests for this acid. The toxicodendric acid of Maisch was thus shown to be acetic acid, and was therefore not the poisonous principle of the plant.
Pfaff obtained the active principle by the following process: The plant was extracted with alcohol, the alcohol was distilled off, and the residue was taken up in ether. The ether solution[Pg 14] was washed with water and dilute sodium carbonate solution, and the ether was evaporated. An oily, black, poisonous substance partly soluble in alcohol was obtained. To get the active principle in a pure state, this residue was extracted with alcohol and filtered and the filtrate was precipitated fractionally by lead acetate. The final precipitates consisted of the lead compound of the poison in a pure state. On decomposing the lead compounds with ammonium sulphide, shaking out with ether, and letting the ether evaporate spontaneously, a non-volatile oil was obtained which gave the characteristic skin eruptions. The pure lead compounds made in different preparations were analyzed and assigned the formula C21H30O4Pb. The oil itself was not analyzed. Pfaff proposed the name Toxicodendrol for the oil. He found that it was not volatile, was decomposed by heat, was soluble in alcohol, ether, chloroform, benzene, etc., but insoluble in water. Its effects upon the human skin were studied in many experiments upon himself and others. It was shown that an exceedingly minute quantity of the poison will produce the dermatitis, even 1/1000 milligram applied in olive oil being active. The oil was given internally to rabbits, its effects being most marked on the kidneys.
The oil obtained by Pfaff from Rhus venenata seemed to be identical with that from Rhus toxicodendron.
The writer's investigation was undertaken with the object of attempting to throw more light on the chemical nature of the poisonous substance found in Rhus toxicodendron. Soon after commencing work, however, it became apparent that the poison could be more intelligently studied if the substances associated with it in the plant were first identified; the scope of the work was therefore extended to an investigation of the other constituents of the plant, and it was hoped that a knowledge of the properties of these constituents would suggest a more economical way of getting the poison than the method of fractional precipitation.
The crude material for this work was prepared by Messrs. Parke, Davis & Co., of Detroit, Mich., according to special instructions submitted to them: 67-1/2 pounds of fresh leaves and flowers of poison ivy were collected near Detroit and carefully inspected by a competent botanist. This material was thoroughly macerated and put into ten-liter bottles with ether. The mass was thoroughly shaken, water being added to make it more[Pg 15] mobile. The ether was then separated off and the extraction was repeated three times in the same way to insure complete removal of the toxicodendrol. The ether extracts were combined, thoroughly dried with anhydrous sodium sulphate, and the ether was distilled off, the temperature being kept below 40° C. during the entire distillation. The residue after the removal of the ether was a thick, black, tar-like mass, weighing 3 pounds 11 ounces. In extracting the plant, about twenty-four gallons of ether were used. It is a significant fact in regard to the volatility of the poison that during the process of preparing this material none of the employees engaged in the work were in any way affected, since proper precautions were taken and the utensils were handled with rubber gloves.
The crude ether extract, which will be designated as the "original material," was shipped to Baltimore in August and was kept in a cool place until November when the investigation was begun. When the bottle was opened, there seemed to be an escape of a vapor, and a nauseating odor suggesting crushed green leaves pervaded the atmosphere. Some days later, irregular red patches appeared on the face though a mask of cotton cloth was worn during the work, and the hands were protected by rubber gloves.
Assuming from Pfaff's work that this original material contained the non-volatile oil toxicodendrol, the first experiment was to try to distil it out under diminished pressure. For this purpose, an Anschütze distilling bulb containing ten grams of the tar was connected with a vacuum pump. After a pressure of 2 mm. had been established the bulb was gradually heated in a bath of Wood's metal. Nothing distilled over. The material began to carbonize at a temperature of 140° to 150°.
It was then thought that perhaps the oil could be converted into an ester which might be more volatile and could be distilled out. 20 grams of the original material were dissolved in 100 cc. of absolute alcohol containing 3 grains of hydrochloric acid gas, and the mixture was heated 10 hours on a water-bath under a return condenser. After the heating, the mixture had a delightful ethereal odor. The flask was corked and left standing several weeks while other work was in progress. The ester solution was then put in a vacuum desiccator over sulphuric acid and the alcohol evaporated. A black, tarry, solid mass was left having the ester odor. It was extracted with warm water and filtered from insoluble tar. The filtrate had a green color and the ethereal odor. It was shaken out with ether; the ether layer had a blood-red color while the water layer was deep green.[Pg 16] The extraction with ether was continued until the water layer was no longer green. The combined ether extracts were evaporated in a desiccator without heat. A black tar-like solid was left very much like the original material, but it had the ester odor. It was partly soluble in water and readily soluble in alcohol. The alcoholic solution was tested on the skin and found to be not poisonous. The ester, or mixture of esters, was not investigated further in this connection, but was later shown to give the reactions for gallic acid and methyl furfurol. These reactions will be referred to in connection with other experiments.
After a few other preliminary experiments, it became evident that the original material was a complex mixture of substances and that it would have to be fractionated by some means and the fractions studied separately.
A portion of the original substance was treated with 50 per cent. alcohol and was found to be partly soluble in this medium. The solution was filtered from insoluble tar. A portion of the yellow filtrate gave a reddish yellow precipitate with lead acetate. The alcoholic solution was distilled in an Anschütze flask under diminished pressure; a yellow liquid condensed in the arm of the flask while most of the alcohol was collected in a bottle connected with the arm. The yellow liquid was acid to litmus. Water was added, the solution was shaken out with ether and the ether was evaporated. When the small residue was completely dry, it was a yellow solid soluble in dilute alcohol and acid to litmus. The substance was not volatile enough to justify the use of this method for getting it.
Chlorophyll could not be removed from the original substance because the solvents for chlorophyll such as alcohol, ether, fats, petroleum, and carbon bisulphide dissolve large quantities of the mixture.
A precipitate obtained by adding lead acetate to a filtered solution of the original substance in 50 per cent. alcohol was suspended in water, decomposed by hydrogen sulphide, shaken out with ether and the ether evaporated. The residue appeared at first to be a yellow oil, but on complete evaporation of the ether in a desiccator, a yellow solid was obtained—apparently the same as that obtained by vacuum distillation.
A solution of the original material in 50 per cent. alcohol was filtered through bone-black and the filtrate was colorless. Examination showed that everything had been removed by the bone-black and the filtrate was apparently pure alcohol and water.[Pg 17]
In precipitating an alcoholic solution of the crude material with a solution of lead acetate, it was noticed that at least two kinds of precipitates were formed. The part that went down first was darker in color than that thrown down later. Pfaff used the last fractions in obtaining his oil and stated that these precipitates consisted of the lead compound of the oil in a pure state. It was found by experiment that the darker part was soluble in ether while the lighter part was not. This indicated that the darker part consisted of tarry matter which was brought down mechanically or separated out when the alcoholic solution was diluted by the lead acetate solution, or was perhaps a lead compound soluble in ether. To test this point an experiment was made as follows: Some of the crude material was thoroughly extracted with 50 per cent. alcohol. The tar insoluble in 50 per cent. alcohol was then treated with 95 per cent. alcohol; most of it dissolved; the solution was filtered and lead acetate in 50 per cent. alcohol was added. A greenish colored precipitate was formed which was filtered off and found to be completely soluble in ether and soluble to a considerable extent in strong alcohol. These experiments suggested that the light colored lead compound which was thought to contain the poison could be purified by extraction with ether in a Soxhlet apparatus more conveniently than by the tedious process of fractional precipitation. Further preliminary experiments showed that 50 per cent. alcohol extracted from the original material all of the substance or substances which gave the light colored precipitate and dissolved only a small amount of the tar.
Two hundred and eighty-eight grams of the crude material were then extracted several times with 50 per cent. alcohol and filtered; the insoluble tar was washed and saved for examination. To the filtrate was added an excess of a solution of lead acetate in 50 per cent. alcohol. The large precipitate, which will be designated as "precipitate A," was filtered and drained by suction in a Büchner funnel. The alcoholic "filtrate A" was saved. Precipitate A was extracted with ether in Soxhlet extractors until the ether came over practically colorless, the operation being interrupted from time to time to stir up the precipitate in the thimble. The green colored ether solution was saved for future examination. The lead precipitate, after extraction with ether and drying, weighed about 116 grams. A portion of this lead compound was decomposed by hydrogen sulphide in a mixture of water and ether which was well shaken during the operation. The ether was separated, filtered, and evaporated under diminished pressure without heat, and there remained a[Pg 18] yellow oily looking residue having a pleasant odor. When the ether and water were completely removed in a vacuum desiccator, a hard, brittle, yellow resin weighing about 16 grams was obtained. It was soluble in alcohol, had a strong acid reaction and was free from nitrogen[16] and sulphur. The nitrogen tests were made by the Lassaign and soda lime methods,[17] and the sulphur test was made with sodium nitroprusside after fusing the residue with sodium. The main portion of the lead compound was decomposed under alcohol by hydrogen sulphide, filtered, and the alcoholic filtrate evaporated in vacuo. The same yellow acid resin was obtained. Experiments continuing through several weeks were made in applying solutions of this resin to rats, rabbits and guinea pigs. Finding it to be without effect upon these animals it was tried on the writer and found to be not poisonous.[18] In the meantime the resin was being studied in the laboratory.
An alcoholic solution of the resin was just neutralized with potassium hydroxide. During the titration, the solution rapidly became dark brown. After neutralization it was shaken with ether; the water solution remained brown while the ether layer was nearly colorless and contained practically no dissolved substance. A portion of the water solution of the potassium salt on being acidified with sulphuric acid and standing over night, deposited a slight precipitate. The solution of the potassium salt gave a heavy precipitate with lead acetate somewhat similar to the original lead precipitate A, and also slight precipitates with salts of zinc, mercury, copper, and silver (with reduction). It gave a bluish-black color with impure ferrous sulphate and a dark color with ferric chloride. It reduced ammoniacal silver nitrate and Fehling solution. These experiments indicated the presence of a tannin compound. An alcoholic solution of the resin gave the same color reactions with iron salts as did the potassium salt. To determine which one of the tannin compounds was present was a matter of some difficulty since the di- and tri-hydroxybenzoic acids have in general the same color reactions. The presence of other plant[Pg 19] substances in the solution also interferes with the color tests, and finally, a substance which gives a blue color with iron salts and one giving a green color may be found together in the same plant.[19] Further tests with a solution of the resin in dilute alcohol, and with a water solution of the acid precipitated by adding sulphuric acid to a solution of the resin in potassium hydroxide, led to the conclusion that the acid is gallic acid. These tests were the following:
(1) Boiling with an excess of potassium hydroxide gave a black substance (tauromelanic acid).
(2) The acid was not precipitated by gelatin.
(3) On addition of potassium cyanide a transitory red color appeared which reappeared on shaking with air.
Gallic acid is distinguished from tannic acid by tests (2) and (3). At later stages in the work the potassium, barium, and sodium salts of gallic acid were obtained, and finally the pure acid was made by decomposing the sodium salt with sulphuric acid and crystallizing from water. A portion of the acid so obtained was further purified by dissolving in absolute alcohol and pouring into absolute ether.[20] The melting point behavior of the acid corresponds with that of gallic acid; it melted with decomposition at about 230°. For further identification, some of the acid was converted into an ester by the following process: it was dissolved in 80 per cent. alcohol, hydrochloric acid gas was passed in, and the solution was heated an hour on the water bath. It was then evaporated to a small bulk, neutralized with barium carbonate and extracted with ether. The ether, on evaporation, left the ester which was crystallized from water and dried in a desiccator over sulphuric acid. The anhydrous ester agreed in melting point (156° to 159°) and other properties with the ester of gallic acid described by Grimaux.[21] For the sake of comparison, an ester was made from gallic acid obtained from another source and the two agreed in properties. A mixture of the two esters melted within the limits given for the ester of gallic acid.
While the tests leading to the identification of gallic acid were being made, another series of experiments was in progress. Eleven and one-half grams of the resin obtained from lead precipitate A by decomposition with hydrogen sulphide were treated with 0.1 n. potassium hydroxide added from a burette until the acid was exactly neutralized. All went into solution.[Pg 20] On shaking with ether, some of the potassium salt separated out and was saved for examination. The solution became brown on exposure to air and got darker as the work proceeded. The acid in solution as a potassium salt was precipitated out in four fractions by adding for each fraction one-fourth the amount of 0.1 n. sulphuric acid required to neutralize the potassium hydroxide used. The precipitates were filtered off and examined. The first was small in amount, gummy and hard to filter. The solution was shaken with ether after each precipitate had been filtered off. The succeeding precipitates were in better condition, but were not pure. All appeared to be impure gallic acid which had become brown by absorption of oxygen. They were saved, however, to be tested for poison. After the last fraction had separated, the filtrate was shaken several times with ether and saved for further examination, which will be described under "Rhamnose." This filtrate is designated as B.
At this stage of the work a portion of the resin obtained from lead precipitate A was tested and found to be not poisonous as already mentioned. By this test, all the substances contained in the lead precipitate A after its extraction with ether in the Soxhlet apparatus, were eliminated from the possible poisonous substances. The poison must therefore have been extracted by the ether.
A fresh portion of the original poisonous material was treated with 50 per cent. alcohol and filtered from insoluble tar. The filtrate was precipitated in six fractions by lead acetate. The last fractions were lighter in color and apparently much purer than the first. The sixth lead precipitate was decomposed by hydrogen sulphide, the light-yellow water solution was tested and found to be not poisonous. It gave the characteristic reactions for gallic acid. The poison, if precipitated at all by lead acetate, must have gone down in one of the preceding fractions. Later experiments showed that it is brought down partly mechanically and partly as a lead compound in the first precipitates.
Having identified gallic acid, and not finding any other phenol derivative in the lead precipitate, some of the original material was extracted with hot water to remove gallic acid and filtered from tar while hot. The filtrate had a deep yellow color. On cooling over night, an olive green precipitate separated out which was dried and found to be a light powder. It was practically[Pg 21] insoluble in cold water, soluble with great difficulty in boiling water from which it separated in yellow flakes, slightly soluble in ether and in acetic acid, but readily soluble in alcohol. The solutions were not acid to litmus, gave a dark color with ferric chloride, an orange-red precipitate with lead acetate which was easily soluble in acetic acid, and an orange-yellow precipitate with stannous chloride. These properties and reactions indicated that the substance was the dye-stuff fisetin and that it occurs in the free state in this plant though it is usually found as a glucoside of fisetin combined with tannic acid. A compound of this kind was found in Rhus cotinus and named "fustin-tannide" by Schmid[22]. He showed that the fustin-tannide could be decomposed by acetic acid into tannic acid and a glucoside, fustin C46H42O21. Fustin, on heating with dilute sulphuric acid, gave fisetin and a sugar supposed to be rhamnose. Fisetin was also found as a glucoside compound in Rhus rhodanthema by Perkin.[23]
The yellow substance which separated from the boiling water solution was further purified by dissolving in a small quantity of hot alcohol and adding hot water. On cooling, the yellow substance separated out in a flocculent condition. Examined under the microscope, the flakes appeared to be made up of masses of fine crystals.
An alcoholic solution of the substance gave a black color with ammonia which became red on addition of more ammonia. Concentrated acids intensified the yellow color of the alcoholic solution. Fehling solution and ammoniacal silver nitrate were reduced by it. Potassium hydroxide added to an alcoholic solution gave at first a deep red color accompanied by a green fluorescence which disappeared, leaving a yellow liquid. With an excess of caustic potash, the red color returned and was permanent. These reactions are characteristic for fisetin.[24]
Furthermore, fisetin should give protocatechuic acid and phloroglucinol by fusion with caustic potash under proper conditions.[25] The experiment was carried out as follows: 2 grams of fisetin were gently heated in a nickel crucible with 6 grams of caustic potash dissolved in 6 cc. water. An inflammable gas, apparently hydrogen, was evolved during the fusion. The pasty mass was dissolved in water, acidified with sulphuric acid, and filtered. The filtrate was shaken out with ether containing one-fourth[Pg 22] its volume of alcohol. The ether was evaporated and the residue was extracted with warm water and filtered. Lead acetate was added to the filtrate to precipitate protocatechuic acid, while phloroglucinol remained in the filtrate from this precipitate. The lead precipitate was suspended in water, decomposed by hydrogen sulphide, filtered, and evaporated to obtain protocatechuic acid. That the substance obtained was protocatechuic acid was shown by the following characteristic tests:
(1) It gave a greenish brown color with ferric chloride; on addition of one drop of a dilute solution of sodium carbonate, the color became dark blue; on adding more sodium carbonate the color became red.
(2) A violet color was obtained when a solution of the acid was treated with a drop of sodium carbonate solution and then with a drop of ferrous sulphate.
(3) It reduced ammoniacal silver nitrate.
(4) It did not reduce Fehling solution.
The filtrate supposed to contain phloroglucinol was treated with hydrogen sulphide to remove lead, filtered, and shaken with ether. The residue left on evaporating the ether was taken up in water. This solution gave the following reactions characteristic for phloroglucinol:
(1) It reduced both silver nitrate and Fehling solution.
(2) It colored pine wood moistened with hydrochloric acid red.
(3) It gave a red color with vanillin and hydrochloric acid, and
(4) A deeper red color with oil of cloves and hydrochloric acid, becoming purple on standing.
(5) It gave a violet color with ferric chloride.
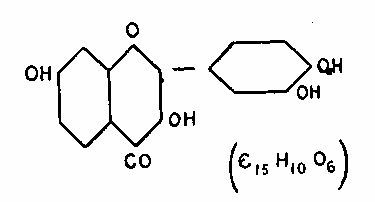
The substance is then, without doubt, fisetin. The formula[26] of fisetin is supposed to be
It was stated above that Schmid obtained a sugar solution by the decomposition of a fisetin-glucoside from Rhus cotinus, and Perkin obtained the same from a glucoside in Rhus rhodanthema. These investigators thought that the sugar was isodulcite or rhamnose, but they did not isolate it on account of the small quantities of material at their disposal. Moreover, the sugar is very hard to crystallize in the presence of other soluble substances and is not found in large quantity in plants. Maquenne[27] could obtain only 15 to 20 gm. of rhamnose by working up 1 kilogram of the berries of Rhamnus infectorius. Assuming that the free fisetin found in poison ivy leaves had its origin in the decomposition of a fisetin-glucoside by natural processes, it was reasonable to suppose that the sugar would also be found in the free state, although, according to Roscoe and Schorlemmer:[28] "Isodulcite does not occur in the free state in nature, but is found as a peculiar ethereal salt belonging to the class of glucosides. On boiling with dilute sulphuric acid, this splits up into isodulcite and other bodies...." The more recent works on the sugars and on plant chemistry[29] mention the occurrence of rhamnose only in the glucoside form, with one possible exception. The exception referred to is the occurrence of a free sugar, supposed to be rhamnose, in a certain palm-wine.[30] Czapek says:[31] "The well-known methyl pentoses do not occur in the free state in plant organisms so far as we know."
Since rhamnose forms a lead compound, the sugar, if present, should be found in the first lead precipitate, A, and also in filtrate A in case it is not completely precipitated in the presence of acetic acid and alcohol.
The filtrate A (about two liters) was examined first. It had a light yellow color, contained an excess of lead acetate, and was acid from the acetic acid liberated in the precipitation of the lead compound A.[32] This filtrate was evaporated to dryness under diminished pressure to remove alcohol, water, and acetic acid. The clear distillate had a peculiar odor suggesting both tea and amyl formate. It was saved for examination and was found to be not poisonous. The residue in the dish after evaporation[Pg 24] was a tough reddish brown, gummy mass which could be drawn out into fine threads. It had a pleasant sweet odor. It was extracted several times with hot water, each portion being filtered. A brownish yellow powder remained undissolved and was saved. The combined filtrates deposited more of the yellow solid on standing. This powder will be referred to later as "P." The filtered liquid was freed from lead by hydrogen sulphide. The solution then had a lemon yellow color, a sweet odor and was acid from acetic acid. On concentrating the solution by evaporation and making a small portion of it alkaline with sodium hydroxide, the yellow color came out very intense[33]. The alkaline solution reduced Fehling solution and ammoniacal silver nitrate, indicating the presence of a sugar. Another portion of the solution gave a slight precipitate with phenyl hydrazine in the cold. The remainder of the solution was evaporated to dryness, extracted with water, filtered, and again evaporated. A dark sticky syrup was left which was only partly soluble in water. This was treated with water, filtered, and the filtrate was evaporated, the water being replaced from time to time to remove acetic acid. Finally the liquid gave the following tests for rhamnose, besides those already mentioned:
(1) With α-naphthol[34] and sulphuric acid, a purple violet color.
(2) With thymol[35] and sulphuric acid, a red color.
(3) With resorcinol[36] and sulphuric acid, red color.
(4) With orcinol[37] and hydrochloric acid, red color.
(5) With ammonium picrate and sodium picrate, yellowish red color.
(6) With phloroglucinol and hydrochloric acid, red color.
(7) It decolorized an alkaline solution of potassium ferricyanide.
(8) It gave a white precipitate with lead acetate.
The filtrate B (p. 20) from which gallic acid was precipitated by sulphuric acid in four fractions was saved to examine for sugar. To remove gallic acid completely, and other vegetable matter, it was shaken out several times with ether, and was kept at a low temperature with salt and ice for a long time. It was left standing for several weeks, during which time more brown matter separated out and was filtered off. The filtrate was[Pg 25] evaporated to a small bulk, cooled, and filtered from crystals of potassium sulphate. The filtrate was evaporated to dryness, the residue taken up in water and filtered through bone-black. Addition of alcohol caused complete precipitation of potassium sulphate. The solution then gave the above mentioned characteristic tests for rhamnose.
All attempts to get the osazone of the sugar by the method of Fischer[38] failed, probably on account of the small quantity of the sugar present. The plant, it will be remembered, was originally extracted with ether in which rhamnose is practically insoluble. The above described tests, however, can leave no doubt as to the identity of the sugar.
Additional evidence that the sugar is rhamnose was obtained by a method described by Maquenne[39] as follows:
"The production of methyl furfurol in the dehydration of isodulcite furnishes a very simple means of characterizing this sugar in mixtures which contain it; it is sufficient, for example, to distil 50 gm. of quercitron wood with as much sulphuric acid and about 150 gm. of water, then to rectify the liquid obtained in order to get several drops of the crude furfurol, which on addition of alcohol and concentrated sulphuric acid gives immediately the green coloration characteristic of methyl furfurol. This procedure is applicable to extracts as well as to entire plants, and has the advantage that it does not require the separation of isodulcite, the crystallization of which is often very slow and at times impossible when it is mixed with other very soluble substances."
The experiment was tried with the crude ether extract of the plant according to the directions of Maquenne, and the green color with alcohol and sulphuric acid was obtained from the thicker oily portion of the distillate. This test can be made with hydrochloric acid[40] as well as with sulphuric. Therefore the color test was tried with the ester mixture prepared in one of the early experiments by boiling the original plant material with hydrochloric acid and alcohol. Methyl furfurol was found here also, this method indeed giving better results than that of Maquenne.
The presence of free rhamnose has thus been shown in the original material, in the first precipitate by lead acetate, and in the filtrate from this precipitate. Experiments to be described under "The Poison" showed that the ether extract from the Soxhlet apparatus contained a substance which yielded rhamnose when hydrolyzed by dilute sulphuric acid.
The presence of free gallic acid, fisetin, and rhamnose in the plant can be readily explained by a series of assumptions for which there is a considerable amount of experimental evidence. There is reason to believe that tannin-like bodies are formed at the expense of chlorophyll,[41] that complex tannin bodies can be broken down by acetic acid (also found in Rhus toxicodendron) into a tannic acid and a glucoside (for example, the "fustin-tannide" mentioned above yields tannic acid and fisetin-glucoside); and finally that the glucoside can be hydrolyzed by acids or enzymes giving, in the sumach plants, fisetin and rhamnose.
Nitrogenous ferments which can effect the hydrolysis of glucosides and give rise to sugars are frequently found in plants, for example, emulsin in almonds, myrosin in mustard, and erythrozym in madder. Acree and Hinkins[42] found that diastase, pancreatin, and a number of other enzymes cause hydrolysis of triacetyl glucose with the formation of glucose and acetic acid. Stevens[43] obtained a nitrogenous oxidizing enzyme from Rhus vernicifera. The close relationship between the poisonous species of Rhus would lead us to suppose that the same soluble ferment exists in poison ivy, though it was not detected in the original material used in these experiments, probably because the plant was extracted with ether in which the enzyme is insoluble. The existence of such a soluble ferment would explain the presence of free sugar and free fisetin.
The brown substance P, obtained from filtrate A by evaporation and extracting the residue with hot water, was suspended in warm water and dilute sulphuric was added. A white precipitate was formed and a strong fatty acid odor was developed. After the mixture had been heated for some hours on the water bath a small portion was made alkaline and it reduced Fehling solution. The main solution was filtered and the precipitate supposed to be a fatty acid was saved. The filtrate was neutralized[Pg 27] with barium carbonate, filtered, evaporated, freed from caramel, and the solution then gave the tests mentioned above for rhamnose.
A portion of the precipitate supposed to be a fatty acid was ignited in a porcelain spoon. It fused, carbonized, and burned. The remainder was heated with alcoholic potash and reprecipitated with hydrochloric acid. The precipitate was washed and heated with alcohol. Part of it dissolved. The insoluble part was found to be a lead compound. On boiling it with hydrochloric acid and cooling, lead chloride crystallized out. This was confirmed by dissolving the lead chloride in hot water and precipitating as lead sulphide. These experiments were not carried farther on account of the small quantity of material, but they show that the gummy substance obtained from filtrate A contained rhamnose (either as a lead compound of free sugar or as a lead compound of a rhamnoside), and also, most probably, the lead compound of an organic acid.[44]
Several times in the course of this work, extracts of the original plant material in alcohol and in water were distilled under diminished pressure for the purpose of concentrating the solutions. The distillate, in every case, had an ethereal odor suggesting amyl formate in very dilute solution, but was more fragrant. The distillate from a water extract was examined. It was a clear liquid, a little darker than pure water, was not poisonous, was neutral to litmus paper, gave no color with ferric chloride, reduced ammoniacal silver nitrate, but not Fehling solution, and gave a faint red color with dilute ammonium hydroxide and with sodium carbonate.
A small quantity of a finely divided black precipitate separated out from the water distillate on standing.
The substance with the fragrant odor was extracted by shaking the distillate with ether and letting the ether evaporate spontaneously. A very small quantity of a yellow solid was deposited on the sides of the dish. This substance had a strong and persistent odor, so sweet as to be almost nauseating. Not enough was obtained for examination or analysis. This fragrant residue was difficultly soluble in water and the solution reduced[Pg 28] silver nitrate in ammonia. A steam distillate of the original plant material had the same fragrant odor as the distillate from a water extract.
288 grams of the original poisonous material were extracted with 50 per cent. alcohol, and this alcoholic solution was precipitated with lead acetate in the manner already described (p. 17). The lead precipitate so obtained was extracted with ether in Soxhlet extractors and after the extraction was found by test to be free from poison. Therefore the poison, if precipitated by the lead acetate, must have been extracted by the ether. This ether solution had a dark green color, and was acid from acetic acid brought down in the lead precipitate. The ether was evaporated in a vacuum desiccator without heat and there remained a small quantity of an acid mixture of water and a soft tar; the watery part was colored green, showing that the tar was soluble to some extent in dilute acetic acid. The mixture had the peculiar odor of the original material. A small drop of the green watery part was applied to the wrist, allowed to remain a few minutes and was then removed by absorbent paper, but the spot was not washed. Itching and reddening of the skin commenced within twenty-four hours. At the end of forty-eight hours, there was a well developed case of poisoning. How this was cured will be described in another place.
A small portion of the poisonous mixture was dissolved in alcohol, and this solution was divided into three parts. The first part was treated with ferric chloride, but it gave no color reaction. Another portion of the alcoholic solution was diluted with water. It became turbid. The third portion gave a dirty-green precipitate with lead acetate, which seemed to come down more readily when the solution was diluted with water. The main portion of the poisonous mixture was then dissolved in 95 per cent. alcohol and lead acetate in 50 per cent. alcohol was added. The precipitate was filtered, washed, and decomposed by hydrogen sulphide in a mixture of water and ether. The ether solution was filtered and evaporated. The residue was a tar which, on standing in a desiccator for some time, became dry enough to break into sticky lumps. An alcoholic solution of this substance gave a dark color with ferric chloride and a light colored precipitate with lead acetate.
To get more of the poisonous tar for study, 233 grams of original material were extracted with 95 per cent. alcohol. Strong alcohol was used in order to dissolve as much of the tar as[Pg 29] possible. The solution had a dark greenish color, but was somewhat yellow in thin layers. The undissolved tar was filtered off and extracted twice again in the same way. The tar left after the third extraction was only slightly soluble in alcohol, and its solution was not poisonous. The three filtrates from these three extractions were precipitated separately by lead acetate in 50 per cent. alcohol. The first precipitate was largest, darkest in color, and carried down more tarry matter. The second was light green, and the third was quite small, black, and was not a lead compound at all, but some of the tar which separated out on diluting the strong alcohol with the weaker grade containing lead acetate. It was soluble in ether and less soluble in alcohol. The alcoholic solution of this third lot gave no precipitate with hydrogen sulphide. The first and second lead precipitates were filtered by suction and washed with water. They were kept a day or two in a desiccator over sulphuric acid, but did not become completely dry. The weight of these two moist precipitates together was 172 grams. They were combined and extracted with ether in Soxhlet extractors which were kept in operation during work hours for three days.
In the meantime the alcoholic filtrates from these lead precipitates were combined and concentrated on the water bath by distilling off two liters of alcohol. The alcohol obtained had the peculiar odor of the original material, but was not poisonous.
After a long extraction of the lead precipitate in the Soxhlet extractors, the green ether solutions were combined and washed by shaking them with water to remove lead acetate and acetic acid in case any should have been held in the lead precipitate. The ether was distilled off at a low temperature and there remained a soft tar, a portion of which was not completely soluble in 95 per cent. alcohol. The alcoholic solution had a greenish yellow color and was poisonous. The tar was also partly soluble in acetic acid, and this solution was found to contain lead. Thinking that the lead acetate had not been completely washed out, the main part of the tar was dissolved in ether and shaken with water. The wash water continued to give a test for lead as long as the washing was continued. This indicated probably the hydrolysis of an unstable lead compound. Hydrogen sulphide was passed into the ether solution mixed with water to remove the lead. Lead sulphide was filtered off, and the ether was evaporated. A small portion of the tar residue in alcoholic solution gave a color reaction with ferric chloride. As this may have been due to traces of lead gallate dissolved in the extraction with ether and afterwards decomposed by hydrogen[Pg 30] sulphide, the main portion of the tar was redissolved in ether and shaken with water until it no longer reacted with ferric chloride. The ether was then evaporated and a soft, black, poisonous tar or gum of uniform consistency was left which was shown by tests to be free from gallic acid and lead. These experiments showed that some of the poison was precipitated as a lead compound soluble in ether and some was brought down mechanically in the free state. To see if the extraction with ether in the Soxhlet apparatus was complete, the residue in the thimbles was decomposed by hydrogen sulphide and shaken with ether. The dark colored ether solution was freed from gallic acid by shaking with water and dilute sodium carbonate solution, and was evaporated. A small quantity of tar was obtained which was added to the main portion.
A solution of the poisonous tar in 95 per cent. alcohol did not reduce Fehling solution and did not give a precipitate with lead acetate except the separation of a small quantity of tar, which was not a lead compound. The lead compound of the poison was apparently soluble in 95 per cent. alcohol as well as in ether, for it would not precipitate in this medium, although it was found in the original precipitate by lead acetate. The alcoholic solution of the tar became turbid on diluting with water.
In order to see if the poison is volatile with vapor of acetic acid, since this acid is found in the plant and it is thought by some that the poison is volatile, a portion of the tar was distilled under diminished pressure with acetic acid. It was soluble to some extent in the acid. The temperature did not go higher than 55° during the distillation. A tube containing cotton wet with sweet oil was placed between the receiver and the water suction so that the uncondensed vapors would have to pass through the cotton. This cotton was rubbed on the skin and was not poisonous. The yellow distillate collected in the receiver was also tested and was not poisonous.
About 5 grams of the poisonous tar free from gallic acid and sugar was dissolved in alcohol, and dilute (2 per cent.) sulphuric acid was added. Some of the tar separated out on diluting the alcohol with the acid. The mixture was heated on a water bath during work hours for four days. A purple and green fluorescent solution was formed, though much tar was left apparently unchanged. The alcohol was evaporated off and the solution was filtered from tar. The fluorescent filtrate was shaken with[Pg 31] ether, by which the green substance was removed, leaving the solution purple. The ether left, on evaporation, a small quantity of a green substance having a pleasant ester odor. It was not further examined. A portion of the purple solution was exactly neutralized with sodium carbonate. This solution gave a blue-black color with ferric chloride which became red on addition of another drop of sodium carbonate, indicating gallic acid. It also reduced Fehling solution.
Another portion of the purple solution was made alkaline with sodium carbonate. A reddish-brown flocculent precipitate was formed and was filtered off. The filtrate did not give any color with ferric chloride, but it reduced Fehling solution. It also gave the test for rhamnose with α-naphthol.
The main portion of the purple solution was made alkaline with sodium carbonate; the precipitate was filtered off and dissolved in acetic acid. This solution was yellow and gave a reaction with ferric chloride similar to that of gallic acid. The filtrate from the precipitate by sodium carbonate was concentrated by evaporation until sodium sulphate began to crystallize out. Alcohol was added to precipitate the sodium sulphate completely, the mixture was heated and filtered. The alcoholic filtrate was concentrated to a syrup which reduced Fehling solution and gave the characteristic tests for rhamnose already described. By this hydrolysis, the tar was split up into rhamnose and some form of gallic acid which could be precipitated by sodium carbonate. This compound, whose acetic acid solution was yellow, probably contained fisetin also. The reason for this last statement will appear from the following experiment:
A portion of the poisonous tar was heated in an open dish with strong acetic acid. The tar seemed to be decomposed to some extent, giving a yellow substance. Acetic acid was added from time to time as it evaporated. After several evaporations, water was added, the mixture was heated to boiling and filtered. This filtrate No. 1 will be mentioned later. The residue in the dish consisted of undecomposed tar and an olive-green flaky substance. This substance was heated with a fresh portion of glacial acetic acid. Water was added, and the mixture was boiled and filtered. The filtrate had a deep yellow color suggesting fisetin. It was shaken out with ethyl acetate which became colored yellow. A portion of the ethyl acetate solution[Pg 32] gave an orange red precipitate with lead acetate showing the presence of fisetin. The ethyl acetate was removed from the remainder of the solution by evaporation and the yellow residue was taken up in alcohol. This alcoholic solution gave the characteristic reactions for fisetin with stannous chloride, with potassium hydroxide, with ferric chloride and with Fehling solution.
Filtrate No. 1 obtained by heating the poisonous tar with acetic acid and hot water as described above was investigated as follows: A portion of it gave a reddish colored precipitate with sodium carbonate as in the case when the tar was hydrolyzed with sulphuric acid. The remainder was nearly neutralized with sodium carbonate and lead acetate was added in excess to remove gallic acid. The excess of lead was removed by sulphuric acid, and the sulphuric acid was removed by barium carbonate. The solution on evaporation reduced Fehling solution to some extent, but a white precipitate was also formed.
In this experiment, gallic acid and fisetin and probably sugar were formed by decomposition of the poisonous gum with acetic acid, the acid found in the plant by Pfaff. The presence of free gallic acid, fisetin and rhamnose in the plant can therefore be explained by the natural hydrolysis of a complex gum or tar or a constituent thereof. The poisonous property is lost in the general rearrangement which takes place during hydrolysis.
The poisonous tar was not hydrolyzed by boiling with a dilute solution of sodium carbonate.
It was found, as has been stated elsewhere, that the lead compound of the poison could not be precipitated in 95 per cent. alcohol. Further experiments, however, showed that on extracting the poisonous gum with 50 per cent. alcohol, a portion of it dissolved, and this solution gave a precipitate with lead acetate which was a true lead compound. The remainder of the purified tar (about 10 gm.) was treated with 50 per cent. alcohol and filtered. Very little dissolved in alcohol of this strength, but on addition of lead acetate in 50 per cent. alcohol to the solution, a light colored precipitate was formed, which became dark on standing. It was filtered off, washed free from lead acetate, decomposed by hydrogen sulphide, and shaken out with ether. The ether left, on evaporation, a yellow resinous substance having a faint odor like garlic. By drying in a desiccator, a small quantity of a solid yellow resin was obtained which was completely soluble in alcohol. A very small drop of this solution applied to the skin on the end of a glass rod which had been drawn out to a point caused an eruption in about thirty-six[Pg 33] hours. Following the nomenclature used by Maisch and Pfaff, this substance will be designated as Toxicodendrin, the ending "in" indicating its glucoside nature.
The filtrate from the lead precipitate just described was freed from the excess of lead acetate by hydrogen sulphide, was tested for poison, and was found to be poisonous, showing that the precipitation by lead acetate was not complete even in 50 per cent. alcohol. On spontaneous evaporation of the solution, a yellow, sweet smelling resin was left.
A portion of the alcoholic solution of the toxicodendrin gave a dark coloration with ferric chloride, did not reduce Fehling solution and was slightly acid to litmus.
To see whether the toxicodendrin could be hydrolyzed, the remainder was dissolved in alcohol and dilute sulphuric acid was added. A fine, white precipitate was formed at once which rose to the surface on standing as a light flocculent substance. The mixture was heated for several days on a water bath, filtered from unhydrolyzed resin and the filtrate was neutralized and concentrated in the way already described. The solution obtained reduced Fehling solution. Not enough was obtained for further sugar tests, but all the hydrolysis experiments point to the conclusion that the poisonous substance is a rhamnoside, and is the source of the sugar in the plant.
The reaction with ferric chloride observed whenever a lead compound of the poison is decomposed by hydrogen sulphide may be explained by the formation of traces of gallic acid or fisetin through the action of the weak acids present.
The supply of purified poisonous tar having been exhausted in the preceding experiments, further study of the active principle is postponed until more can be prepared. It is highly desirable to investigate the white precipitate formed by addition of sulphuric acid to an alcoholic solution of the toxicodendrin.
When the purified poisonous material (p. 32) was extracted with 50 per cent. alcohol, only a small quantity was dissolved as was stated above. The insoluble residue was treated with fuming nitric acid. Violent reaction took place at once with copious evolution of red fumes and heat. When the reaction was over, a sticky red gummy mass was left which was slightly soluble in cold water and readily soluble in warm alcohol. The water extract was yellow, and the alcoholic solution was red. That the water extract contained picric acid was shown by the following experiments:[Pg 34]
(1) A portion was gently warmed with a few drops of a strong solution of potassium cyanide and two drops of sodium hydroxide. The red color of potassium isopurpurate was formed.
(2) A portion of the water solution was heated with glucose and a few drops of sodium hydroxide. The deep red color of picraminic acid was produced.
(3) A few drops of an ammoniacal solution of copper sulphate was added to the water extract. A yellow-green precipitate was formed.
(4) The water extract dyed silk, but did not dye cotton cloth.
About 25 gm. of the tar left after extracting the original material with hot water was dissolved in ether and poured into a glass retort containing soda lime. The ether was distilled out, leaving the tar intimately mixed with the soda lime. The retort was then gradually heated. Vapors and liquid were given off, both of which turned red litmus blue and had a strong odor like tobacco smoke. No odor of ammonia was detected.[45] At the high temperature of the triple burner, a semi-solid, red, greasy substance collected in and closed the condenser tube. This substance had the same powerful odor as the liquid portion of the distillate. The clear, watery portion of the distillate was separated from the thicker parts, and was found to contain pyrrol and pyridine derivatives by the following characteristic tests:
(1) Wood moistened with hydrochloric acid was turned red by it.
(2) Colorless fumes were formed when brought near hydrochloric acid; mixed with hydrochloric acid, a red insoluble substance was formed.
(3) It precipitated the hydroxides of iron, gave a light blue precipitate with copper sulphate, and a white precipitate with mercuric chloride.
The greasy, semi-solid mass was extracted with 10 per cent. hydrochloric acid and filtered. On addition of a solution of mercuric chloride to the red filtrate, a brown flocculent precipitate was formed. It was filtered off and distilled with caustic soda, but the distillate did not contain pyridine.
In the early stages of this work some experiments were made to see if potassium permanganate could be used to purify the lead precipitate by oxidizing the tar brought down in precipitation. It was found that the permanganate attacked the lead precipitate as well as the other organic matter in the vessel. This fact and the well-known value of permanganate in treating skin diseases, its use as an antidote for some kinds of alkaloid poisoning,[47] as an antidote given to cattle poisoned by plants,[48] and as an antidote for snake bites,[49] suggested its use as a remedy for Rhus poisoning. Maisch[50] mentioned that he had used it with success, but it never came into general use, probably on account of its staining the skin and clothing. In carrying out this work abundant opportunities for testing its value as a remedy for the dermatitis caused by poison ivy were afforded by many cases of accidental and intentional poisoning. The best example of the latter was obtained with the ether solution from the extraction of the lead precipitate in the Soxhlet apparatus (page 28). After removing the ether, a small drop of the residue was applied to the wrist as described. An itching red spot about the size of a dime was noticed in thirty-six hours, and it steadily increased in size. Nearly two days after the application of the poison, a dilute solution of potassium permanganate containing a little caustic potash was rubbed into the spot until the pimples were destroyed. A little black spot was left wherever there had been a pimple, showing that the permanganate had been reduced to oxide in the skin. The place was washed and nothing more was thought of it until the morning following, when it was noticed that the wrist had commenced to swell during the night, and the characteristic watery secretion was running from the poisoned spot. More permanganate solution was applied without potash and the wrist was bandaged, thinking that this would prevent the spreading of the eruption, but it really facilitated spreading by becoming saturated with the poisonous fluid and keeping it in contact with a larger surface of skin. In the meantime the swelling and inflammation had extended nearly to the elbow. The arm now had the appearance of having been bitten by a[Pg 36] snake. To reduce the swelling it was immersed in hot water. This seemed to bring out the eruption very quickly and the blisters were treated with permanganate as fast as they appeared. The swelling was reduced, but returned during the night. On the evening following, the forearm was immersed in a bowl of hot permanganate solution containing a little caustic potash. The solution was kept as hot as could be borne for about half an hour. After this bath, the poison was completely oxidized, for the swelling was reduced and did not return, nor was there any fresh eruption. What appeared to be a severe case of poisoning was thus cured very quickly. The use of hot water not only reduces the swelling, but also helps to destroy the poison. The action of permanganate is also more rapid at high temperatures.
The oxidizing power of permanganate, as is well known, is greater in acid solution than in alkaline, five atoms of oxygen being available in the former and three in the latter, according to these equations:
Permanganate was used as a remedy in some cases mixed with dilute sulphuric acid, and in others, with zinc sulphate; also with lime water. It was found to be satisfactory whether used alone or with any of the substances mentioned, provided it was well rubbed into the skin. The concentration of the solution used was varied according to the location and condition of the eruption. Where the skin was thin or already broken, dilute solutions (one per cent.) were used. In one case, the eruption appeared in the palm of the hand where the skin was so thick that it was necessary to open it before the remedies could reach the poison. The difficulty of getting the remedy in contact with the poison in the skin is the reason why the eruption is hard to cure.
The remedy most commonly used for this eruption is an alcoholic solution of lead acetate. This remedy is unsatisfactory for the reason that its action consists in depositing an unstable lead compound of the poison in the skin where the conditions of moisture and temperature are favorable for its decomposition, liberating the poison with all its irritant properties. Moreover, alcoholic preparations should not be used because the alcohol dissolves the poison and, on evaporation, leaves it spread over a larger surface like a varnish. Potassium permanganate, however, oxidizes the poison completely. The only objection to the[Pg 37] use of permanganate of which the writer is aware is that it stains the skin. The stain can be removed by vigorous scrubbing with soap, or it will wear off gradually in a few days. It can be removed at once by certain acids, but these should not be used by persons not familiar with their action.
With the knowledge of the facts mentioned, many solutions were tested for poison by applying them to the skin, and when an eruption appeared, it was cured quickly and permanently by rubbing in a permanganate solution, usually mixed with dilute sulphuric acid.
[16] Nitrogen was found very readily by the soda lime test in the tar left after extracting the original material with 50 per cent. alcohol, but was not found by the Lassaign test.
[17] Stevens. Amer. Jour. Pharm. 77, 255, June, 1905.
[18] Whenever it is stated in this paper that a solution was poisonous or not poisonous, the test was made by the writer upon himself.
[19] Liebig's Annalen, CXI, p. 215.
[20] Über Mategerbstoff, p. 20.
[21] Bull. Soc. Chim. (II), Vol. 2, 95 (1864).
[22] Berichte 19, 1735 (1886).
[23] Jour. Chem. Soc. 71, 1194 (1897).
[24] Berichte 19, 1740.
[25] Ibid. 1747; Annalen, 112, 97.
[26] Biochem. Pflan. II, 521.
[27] Ann. de Chim. et de Phys., 6th Series, XXII, 76 (1891).
[28] Treatise on Chem., Vol. III, Pt. III, 492.
[29] Les Sucres; Chem. der Zuck.; Biochem. der Pflan.
[30] Chem. Zeit. 23, Rep. 177.
[31] Loc. cit. 1, 209.
[32] On standing several weeks, a small quantity of tar separated out on the walls of the vessel, also a brown precipitate which was filtered off, suspended in water, and hydrogen sulphide was being passed in when an accident occurred and it was lost.
[33] "By warming with alkalies or barium hydroxide, rhamnose is colored yellow." Chem. der Zuck. I, 177.
[34] Ibid. 188.
[35] Ibid.
[36] Rayman, Sur L'Isodulcite, Bull. Soc. Chim. 47, 668 (1887).
[37] Acides Gummiques.
[38] Berichte XX, pp. 1089, 1091, 1188, 2566.
[39] Ann. de Chim. et de Phys. (6) XXII, 93 (1891).
[40] Biochem. der Pflan. I, 210.
[41] Comptes rendus CXV, 892.
[42] Amer. Chem. Jour. 28, 370.
[43] Amer. Jour. Pharm. 77, 255 (June, 1905); 78, 53 (Feb., 1906).
[44] A wax obtained from Rhus succedanea was shown by Stahmer to contain palmitic acid and glycerol in the form of glycerol palmitate. Annalen 43, 343, (1842).
[45] See Amer. Jour. Pharm. 77, 256.
[46] This section is added in the hope that it may be of use to others who are subject to this form of poisoning.
[47] Moor, N. Y. Med. Rec. 45 (1894), 200.
[48] Bull. No. 26, U. S. Dept. Agr., Div. of Bot. 47.
[49] Lacerda, Comptes rendus 93 (1881) 466-469.
[50] Amer. Jour. Med. Sci. 52 (1866), 285.
Leaves and flowers of the poison ivy plant were extracted with ether and the ether was removed by evaporation. In the residue, the following substances were found and studied: gallic acid, fisetin, the sugar rhamnose, and a poisonous tar, gum, or wax.
The lead compound of the poison was soluble in ether; this fact gave a means of separating the poisonous substance from the non-poisonous matter in one operation.
The poison was not volatile with vapor of acetic acid, or with vapor of alcohol.
The poisonous tar or wax was decomposed by acids and yielded gallic acid, fisetin, and rhamnose, showing the probable source of these compounds in the plant, and indicating that the poison is a complex substance of a glucoside nature.
It was found that a portion of the poisonous substance could be precipitated by lead acetate from a solution of the purified tar in 50 per cent. alcohol.
All cases of poisoning developed on the writer were easily cured with potassium permanganate.
The following method is suggested for obtaining the poisonous substance from the plant: Extract the plant with alcohol, filter, and precipitate at once with lead acetate. Wash the precipitate, dry, and extract with ether in Soxhlet extractors (loosely filled). Combine the ether extracts, mix with water, and pass in hydrogen sulphide. Separate the water and the ether solution, and filter the latter. Wash the ether solution thoroughly by shaking with water, and then evaporate at a low temperature.
William Anderson Syme, the author of this dissertation, was born in Raleigh, N. C., on July 11, 1879. He was prepared for college at the Raleigh Male Academy, entered the North Carolina College of Agriculture and Mechanic Arts in 1896, and was graduated in 1899 with the degree B. S. He was an Instructor in Chemistry at the same College from January 1st, 1900, until June, 1903, when he received the degree M. S. for graduate work. In October following, he entered Johns Hopkins University as a graduate student in Chemistry, and was awarded one of the North Carolina Scholarships. His minor subjects are Physical Chemistry and Biology.
End of the Project Gutenberg EBook of Some Constituents of the Poison Ivy
Plant: (Rhus Toxicodendron), by William Anderson Syme
*** END OF THIS PROJECT GUTENBERG EBOOK SOME CONSTITUENTS--POISON IVY PLANT ***
***** This file should be named 34510-h.htm or 34510-h.zip *****
This and all associated files of various formats will be found in:
https://www.gutenberg.org/3/4/5/1/34510/
Produced by Bryan Ness, Josephine Paolucci and the Online
Distributed Proofreading Team at https://www.pgdp.net. (This
book was produced from scanned images of public domain
material from the Google Print project.)
Updated editions will replace the previous one--the old editions
will be renamed.
Creating the works from public domain print editions means that no
one owns a United States copyright in these works, so the Foundation
(and you!) can copy and distribute it in the United States without
permission and without paying copyright royalties. Special rules,
set forth in the General Terms of Use part of this license, apply to
copying and distributing Project Gutenberg-tm electronic works to
protect the PROJECT GUTENBERG-tm concept and trademark. Project
Gutenberg is a registered trademark, and may not be used if you
charge for the eBooks, unless you receive specific permission. If you
do not charge anything for copies of this eBook, complying with the
rules is very easy. You may use this eBook for nearly any purpose
such as creation of derivative works, reports, performances and
research. They may be modified and printed and given away--you may do
practically ANYTHING with public domain eBooks. Redistribution is
subject to the trademark license, especially commercial
redistribution.
*** START: FULL LICENSE ***
THE FULL PROJECT GUTENBERG LICENSE
PLEASE READ THIS BEFORE YOU DISTRIBUTE OR USE THIS WORK
To protect the Project Gutenberg-tm mission of promoting the free
distribution of electronic works, by using or distributing this work
(or any other work associated in any way with the phrase "Project
Gutenberg"), you agree to comply with all the terms of the Full Project
Gutenberg-tm License (available with this file or online at
https://gutenberg.org/license).
Section 1. General Terms of Use and Redistributing Project Gutenberg-tm
electronic works
1.A. By reading or using any part of this Project Gutenberg-tm
electronic work, you indicate that you have read, understand, agree to
and accept all the terms of this license and intellectual property
(trademark/copyright) agreement. If you do not agree to abide by all
the terms of this agreement, you must cease using and return or destroy
all copies of Project Gutenberg-tm electronic works in your possession.
If you paid a fee for obtaining a copy of or access to a Project
Gutenberg-tm electronic work and you do not agree to be bound by the
terms of this agreement, you may obtain a refund from the person or
entity to whom you paid the fee as set forth in paragraph 1.E.8.
1.B. "Project Gutenberg" is a registered trademark. It may only be
used on or associated in any way with an electronic work by people who
agree to be bound by the terms of this agreement. There are a few
things that you can do with most Project Gutenberg-tm electronic works
even without complying with the full terms of this agreement. See
paragraph 1.C below. There are a lot of things you can do with Project
Gutenberg-tm electronic works if you follow the terms of this agreement
and help preserve free future access to Project Gutenberg-tm electronic
works. See paragraph 1.E below.
1.C. The Project Gutenberg Literary Archive Foundation ("the Foundation"
or PGLAF), owns a compilation copyright in the collection of Project
Gutenberg-tm electronic works. Nearly all the individual works in the
collection are in the public domain in the United States. If an
individual work is in the public domain in the United States and you are
located in the United States, we do not claim a right to prevent you from
copying, distributing, performing, displaying or creating derivative
works based on the work as long as all references to Project Gutenberg
are removed. Of course, we hope that you will support the Project
Gutenberg-tm mission of promoting free access to electronic works by
freely sharing Project Gutenberg-tm works in compliance with the terms of
this agreement for keeping the Project Gutenberg-tm name associated with
the work. You can easily comply with the terms of this agreement by
keeping this work in the same format with its attached full Project
Gutenberg-tm License when you share it without charge with others.
1.D. The copyright laws of the place where you are located also govern
what you can do with this work. Copyright laws in most countries are in
a constant state of change. If you are outside the United States, check
the laws of your country in addition to the terms of this agreement
before downloading, copying, displaying, performing, distributing or
creating derivative works based on this work or any other Project
Gutenberg-tm work. The Foundation makes no representations concerning
the copyright status of any work in any country outside the United
States.
1.E. Unless you have removed all references to Project Gutenberg:
1.E.1. The following sentence, with active links to, or other immediate
access to, the full Project Gutenberg-tm License must appear prominently
whenever any copy of a Project Gutenberg-tm work (any work on which the
phrase "Project Gutenberg" appears, or with which the phrase "Project
Gutenberg" is associated) is accessed, displayed, performed, viewed,
copied or distributed:
This eBook is for the use of anyone anywhere at no cost and with
almost no restrictions whatsoever. You may copy it, give it away or
re-use it under the terms of the Project Gutenberg License included
with this eBook or online at www.gutenberg.org
1.E.2. If an individual Project Gutenberg-tm electronic work is derived
from the public domain (does not contain a notice indicating that it is
posted with permission of the copyright holder), the work can be copied
and distributed to anyone in the United States without paying any fees
or charges. If you are redistributing or providing access to a work
with the phrase "Project Gutenberg" associated with or appearing on the
work, you must comply either with the requirements of paragraphs 1.E.1
through 1.E.7 or obtain permission for the use of the work and the
Project Gutenberg-tm trademark as set forth in paragraphs 1.E.8 or
1.E.9.
1.E.3. If an individual Project Gutenberg-tm electronic work is posted
with the permission of the copyright holder, your use and distribution
must comply with both paragraphs 1.E.1 through 1.E.7 and any additional
terms imposed by the copyright holder. Additional terms will be linked
to the Project Gutenberg-tm License for all works posted with the
permission of the copyright holder found at the beginning of this work.
1.E.4. Do not unlink or detach or remove the full Project Gutenberg-tm
License terms from this work, or any files containing a part of this
work or any other work associated with Project Gutenberg-tm.
1.E.5. Do not copy, display, perform, distribute or redistribute this
electronic work, or any part of this electronic work, without
prominently displaying the sentence set forth in paragraph 1.E.1 with
active links or immediate access to the full terms of the Project
Gutenberg-tm License.
1.E.6. You may convert to and distribute this work in any binary,
compressed, marked up, nonproprietary or proprietary form, including any
word processing or hypertext form. However, if you provide access to or
distribute copies of a Project Gutenberg-tm work in a format other than
"Plain Vanilla ASCII" or other format used in the official version
posted on the official Project Gutenberg-tm web site (www.gutenberg.org),
you must, at no additional cost, fee or expense to the user, provide a
copy, a means of exporting a copy, or a means of obtaining a copy upon
request, of the work in its original "Plain Vanilla ASCII" or other
form. Any alternate format must include the full Project Gutenberg-tm
License as specified in paragraph 1.E.1.
1.E.7. Do not charge a fee for access to, viewing, displaying,
performing, copying or distributing any Project Gutenberg-tm works
unless you comply with paragraph 1.E.8 or 1.E.9.
1.E.8. You may charge a reasonable fee for copies of or providing
access to or distributing Project Gutenberg-tm electronic works provided
that
- You pay a royalty fee of 20% of the gross profits you derive from
the use of Project Gutenberg-tm works calculated using the method
you already use to calculate your applicable taxes. The fee is
owed to the owner of the Project Gutenberg-tm trademark, but he
has agreed to donate royalties under this paragraph to the
Project Gutenberg Literary Archive Foundation. Royalty payments
must be paid within 60 days following each date on which you
prepare (or are legally required to prepare) your periodic tax
returns. Royalty payments should be clearly marked as such and
sent to the Project Gutenberg Literary Archive Foundation at the
address specified in Section 4, "Information about donations to
the Project Gutenberg Literary Archive Foundation."
- You provide a full refund of any money paid by a user who notifies
you in writing (or by e-mail) within 30 days of receipt that s/he
does not agree to the terms of the full Project Gutenberg-tm
License. You must require such a user to return or
destroy all copies of the works possessed in a physical medium
and discontinue all use of and all access to other copies of
Project Gutenberg-tm works.
- You provide, in accordance with paragraph 1.F.3, a full refund of any
money paid for a work or a replacement copy, if a defect in the
electronic work is discovered and reported to you within 90 days
of receipt of the work.
- You comply with all other terms of this agreement for free
distribution of Project Gutenberg-tm works.
1.E.9. If you wish to charge a fee or distribute a Project Gutenberg-tm
electronic work or group of works on different terms than are set
forth in this agreement, you must obtain permission in writing from
both the Project Gutenberg Literary Archive Foundation and Michael
Hart, the owner of the Project Gutenberg-tm trademark. Contact the
Foundation as set forth in Section 3 below.
1.F.
1.F.1. Project Gutenberg volunteers and employees expend considerable
effort to identify, do copyright research on, transcribe and proofread
public domain works in creating the Project Gutenberg-tm
collection. Despite these efforts, Project Gutenberg-tm electronic
works, and the medium on which they may be stored, may contain
"Defects," such as, but not limited to, incomplete, inaccurate or
corrupt data, transcription errors, a copyright or other intellectual
property infringement, a defective or damaged disk or other medium, a
computer virus, or computer codes that damage or cannot be read by
your equipment.
1.F.2. LIMITED WARRANTY, DISCLAIMER OF DAMAGES - Except for the "Right
of Replacement or Refund" described in paragraph 1.F.3, the Project
Gutenberg Literary Archive Foundation, the owner of the Project
Gutenberg-tm trademark, and any other party distributing a Project
Gutenberg-tm electronic work under this agreement, disclaim all
liability to you for damages, costs and expenses, including legal
fees. YOU AGREE THAT YOU HAVE NO REMEDIES FOR NEGLIGENCE, STRICT
LIABILITY, BREACH OF WARRANTY OR BREACH OF CONTRACT EXCEPT THOSE
PROVIDED IN PARAGRAPH 1.F.3. YOU AGREE THAT THE FOUNDATION, THE
TRADEMARK OWNER, AND ANY DISTRIBUTOR UNDER THIS AGREEMENT WILL NOT BE
LIABLE TO YOU FOR ACTUAL, DIRECT, INDIRECT, CONSEQUENTIAL, PUNITIVE OR
INCIDENTAL DAMAGES EVEN IF YOU GIVE NOTICE OF THE POSSIBILITY OF SUCH
DAMAGE.
1.F.3. LIMITED RIGHT OF REPLACEMENT OR REFUND - If you discover a
defect in this electronic work within 90 days of receiving it, you can
receive a refund of the money (if any) you paid for it by sending a
written explanation to the person you received the work from. If you
received the work on a physical medium, you must return the medium with
your written explanation. The person or entity that provided you with
the defective work may elect to provide a replacement copy in lieu of a
refund. If you received the work electronically, the person or entity
providing it to you may choose to give you a second opportunity to
receive the work electronically in lieu of a refund. If the second copy
is also defective, you may demand a refund in writing without further
opportunities to fix the problem.
1.F.4. Except for the limited right of replacement or refund set forth
in paragraph 1.F.3, this work is provided to you 'AS-IS' WITH NO OTHER
WARRANTIES OF ANY KIND, EXPRESS OR IMPLIED, INCLUDING BUT NOT LIMITED TO
WARRANTIES OF MERCHANTIBILITY OR FITNESS FOR ANY PURPOSE.
1.F.5. Some states do not allow disclaimers of certain implied
warranties or the exclusion or limitation of certain types of damages.
If any disclaimer or limitation set forth in this agreement violates the
law of the state applicable to this agreement, the agreement shall be
interpreted to make the maximum disclaimer or limitation permitted by
the applicable state law. The invalidity or unenforceability of any
provision of this agreement shall not void the remaining provisions.
1.F.6. INDEMNITY - You agree to indemnify and hold the Foundation, the
trademark owner, any agent or employee of the Foundation, anyone
providing copies of Project Gutenberg-tm electronic works in accordance
with this agreement, and any volunteers associated with the production,
promotion and distribution of Project Gutenberg-tm electronic works,
harmless from all liability, costs and expenses, including legal fees,
that arise directly or indirectly from any of the following which you do
or cause to occur: (a) distribution of this or any Project Gutenberg-tm
work, (b) alteration, modification, or additions or deletions to any
Project Gutenberg-tm work, and (c) any Defect you cause.
Section 2. Information about the Mission of Project Gutenberg-tm
Project Gutenberg-tm is synonymous with the free distribution of
electronic works in formats readable by the widest variety of computers
including obsolete, old, middle-aged and new computers. It exists
because of the efforts of hundreds of volunteers and donations from
people in all walks of life.
Volunteers and financial support to provide volunteers with the
assistance they need are critical to reaching Project Gutenberg-tm's
goals and ensuring that the Project Gutenberg-tm collection will
remain freely available for generations to come. In 2001, the Project
Gutenberg Literary Archive Foundation was created to provide a secure
and permanent future for Project Gutenberg-tm and future generations.
To learn more about the Project Gutenberg Literary Archive Foundation
and how your efforts and donations can help, see Sections 3 and 4
and the Foundation web page at https://www.pglaf.org.
Section 3. Information about the Project Gutenberg Literary Archive
Foundation
The Project Gutenberg Literary Archive Foundation is a non profit
501(c)(3) educational corporation organized under the laws of the
state of Mississippi and granted tax exempt status by the Internal
Revenue Service. The Foundation's EIN or federal tax identification
number is 64-6221541. Its 501(c)(3) letter is posted at
https://pglaf.org/fundraising. Contributions to the Project Gutenberg
Literary Archive Foundation are tax deductible to the full extent
permitted by U.S. federal laws and your state's laws.
The Foundation's principal office is located at 4557 Melan Dr. S.
Fairbanks, AK, 99712., but its volunteers and employees are scattered
throughout numerous locations. Its business office is located at
809 North 1500 West, Salt Lake City, UT 84116, (801) 596-1887, email
business@pglaf.org. Email contact links and up to date contact
information can be found at the Foundation's web site and official
page at https://pglaf.org
For additional contact information:
Dr. Gregory B. Newby
Chief Executive and Director
gbnewby@pglaf.org
Section 4. Information about Donations to the Project Gutenberg
Literary Archive Foundation
Project Gutenberg-tm depends upon and cannot survive without wide
spread public support and donations to carry out its mission of
increasing the number of public domain and licensed works that can be
freely distributed in machine readable form accessible by the widest
array of equipment including outdated equipment. Many small donations
($1 to $5,000) are particularly important to maintaining tax exempt
status with the IRS.
The Foundation is committed to complying with the laws regulating
charities and charitable donations in all 50 states of the United
States. Compliance requirements are not uniform and it takes a
considerable effort, much paperwork and many fees to meet and keep up
with these requirements. We do not solicit donations in locations
where we have not received written confirmation of compliance. To
SEND DONATIONS or determine the status of compliance for any
particular state visit https://pglaf.org
While we cannot and do not solicit contributions from states where we
have not met the solicitation requirements, we know of no prohibition
against accepting unsolicited donations from donors in such states who
approach us with offers to donate.
International donations are gratefully accepted, but we cannot make
any statements concerning tax treatment of donations received from
outside the United States. U.S. laws alone swamp our small staff.
Please check the Project Gutenberg Web pages for current donation
methods and addresses. Donations are accepted in a number of other
ways including including checks, online payments and credit card
donations. To donate, please visit: https://pglaf.org/donate
Section 5. General Information About Project Gutenberg-tm electronic
works.
Professor Michael S. Hart was the originator of the Project Gutenberg-tm
concept of a library of electronic works that could be freely shared
with anyone. For thirty years, he produced and distributed Project
Gutenberg-tm eBooks with only a loose network of volunteer support.
Project Gutenberg-tm eBooks are often created from several printed
editions, all of which are confirmed as Public Domain in the U.S.
unless a copyright notice is included. Thus, we do not necessarily
keep eBooks in compliance with any particular paper edition.
Most people start at our Web site which has the main PG search facility:
https://www.gutenberg.org
This Web site includes information about Project Gutenberg-tm,
including how to make donations to the Project Gutenberg Literary
Archive Foundation, how to help produce our new eBooks, and how to
subscribe to our email newsletter to hear about new eBooks.